5 计算资源及编程
5.1 硬件配置
理论上在个人Windows电脑上面做生物信息学数据分析是不实际的,因为太多的生物信息学相关软件的开发者对windows并不熟练,没办法提供完善的基于windows操作系统的软件。 而且个人Windows电脑配置肯定不会太高,一般的组学测序数据都是10~500G一个样本,而且很多软件运行的时候对内存要求很高,最后这些数据的分析过程会非常耗时,个人电脑在硬盘,内存,cpu方面均不足以承担这个重任。
所以一般建议使用配置比较高的服务器,而且建议给服务器安装linux系统,ubuntu及centos均可。
- 单人使用,人民币2万以内,可以配置16线程+64G内存+4T硬盘
- 1到5人课题组,人民币10~50万,可以配置64线程+512G内存+64T硬盘
- 5人以上的课题组,一般是学校的超算中心有专门的IT来负责服务器。
服务器主要用来做计算,数据分析的时候使用,并不需要直接接触它。所以大家会用个人电脑来远程登录到服务器,在上面执行各种各样的数据处理命令。 如果是windows电脑,那么建议安装winscp+xshell来连接服务器。 如果是MAC电脑,建议用自带的终端即可,还可以用FileZilla软件进行文件传输。
如果是超算中心提供账号即可使用,不需要看攻略了,自然会有专门的对应的培训。 如果是1到5人课题组,找到联想,IBM等商家自然会拿到详细报价,甚至他们会上门进行ppt讲解。 那么需要在本文详细讲解的就是个人服务器,预算2万左右,改如何配置。
生信领域所涉及的计算往往是非持续性的,我相对较熟悉的RNASeq中计算量较大的就是比对步骤了,而比对往往只需要一次就可以! 这导致配备了一台豪华服务器使用率缺很低,用更少的钱做更多的事,配置一台可用于生物分析的PC机! 这篇配置适用于生物信息实验室、学校、研究所这样的单位,没有专业机房和运维人员,服务器使用率不高,经费有限等请场景下,不差钱的豪门请回避。
以下数据来自2017年6月22日京东数据
| 配件 | 配置 | 单价 |
|---|---|---|
| 主板 | 微星(MSI)X99A RAIDER 主板 (Intel X99/LGA2011-3) | 2299 |
| CPU | 英特尔(Intel)Extreme系列 酷睿八核i7-6900K 2011-V3 | 7699 |
| 内存 | 金士顿(Kingston)骇客神条 Fury系列 DDR4 2400 16G | 999*8=7992 |
| 电源 | 安钛克(Antec)额定650W EAG650 PRO 模组电源 | 649 |
| 散热器 | 九州风神(DEEPCOOL)大霜塔 CPU散热器 | 219 |
| 硬盘 | 西部数据(WD)红盘 8TB SATA6Gb/s 128Mb | 2999 |
| 机箱 | 酷冷至尊(CoolerMaster)特警342U3版 | 209 |
总价
| 配置 | 总价 | 优缺点 |
|---|---|---|
| 8核128G内存8T存储 | 22066 | 适合小基因组de novo分析,有参比对分析,主要针对的是de novo需要大内存 |
| 6核128G内存8T存储 | 18666 | 比上一套速度慢些,性价比较高,适用于数据不多情况 |
| 8核64G内存8T存储 | 18070 | 常规分析+小数据存储 |
| 8核64G内存2T存储 | 15520 | 常规分析,存储能力几乎没有 |
| 6核64G内存2T存储 | 12120 | 小数据分析,会有速度影响不过影响不大 |
| 10核128G内存8T存储 | 29366 | 速度相对快一些,性价比较低,比上不足比下有余 |
其中存储是独立于服务器配置的,取决于课题组项目的多少,可以进行按需扩容,下面给出一个42T的raid5磁盘阵列的配置方案,可用于普通主板,配置简单,适用于数据备份存储! 磁盘阵列,麦沃(MAIWO)K8FSAS 全铝 八盘位磁盘阵列柜 单价4999 硬盘选择希捷(SEAGATE)酷鹰系列 6TB 7200转256M总计1599*8=12792 最后总价是17791,当然,大部分实验室可能并没有这么多的数据,不需要配置这个存储。
5.2 软件安装
大部分的数据分析最重要的就是学习使用各种各样的软件了,一般生物信息学软件发布的时候会提供多种种形式以供下载,比如sratoolkit
sratoolkit.2.6.3-centos_linux64.tar.gz 2016-05-25 17:24 61M
sratoolkit.2.6.3-mac64.tar.gz 2016-05-25 17:25 52M
sratoolkit.2.6.3-ubuntu64.tar.gz 2016-05-25 17:25 61M
sratoolkit.2.6.3-win64.zip 2016-05-25 17:23 27M 又或者 NCBI的 blast
ncbi-blast-2.6.0+-1.x86_64.rpm 172 MB
ncbi-blast-2.6.0+-src.tar.gz 19.1 MB
ncbi-blast-2.6.0+-src.zip 22.3 MB
ncbi-blast-2.6.0+-win64.exe 79.7 MB
ncbi-blast-2.6.0+-x64-linux.tar.gz 212 MB
ncbi-blast-2.6.0+-x64-macosx.tar.gz 122 MB
ncbi-blast-2.6.0+-x64-win64.tar.gz 79.5 MB
ncbi-blast-2.6.0+.dmg 123 MB 可以看到软件开发单位提供的有src后缀的源代码文件,还有适用于各个操作系统的预编译版本
5.2.1 二进制软件(预编译版本)
作为新手,建议大家直接根据自己的系统下载预编译版本软件,并且直接解压就可以使用啦。 例子如下:
cd ~/biosoft
mkdir sratoolkit && cd sratoolkit
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.6.3/sratoolkit.2.6.3-centos_linux64.tar.gz
tar zxvf sratoolkit.2.6.3-centos_linux64.tar.gz
~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastdump -h ## 我的系统是linux,所以用上面的代码软件就安装成功可以使用啦,是不是非常简单呢。
5.2.2 源码软件
一般的开源软件发布的时候肯定会把源代码放出来,如果是在linux系统下以源代码方式安装软件,那么一般自己的linux系统要有gcc编译器,还需要有一些库文件,这也是大多数新手被坑的地方。
源代码安装三部曲是:
- step1:配置 ./configure
- step2:编译 make
- step3:安装 sudo make install
这个时候就需要对计算机的操作系统有一定的了解了,比如第一个步骤可以设置–prefiex=安装路径,参数指定软件编译后的可执行文件放在具体哪个路径下,默认的路径需要有root权限。 而第二步经常会遇到的库文件缺失,比如安装bwa软件的zlib,安装samtools的 等等。 总之遇到的坑越多,学到的知识越多,只是对初学者来说,这些知识点是否有必要学习,是否应该这么早学习这些。 如果直接用bioconda来管理生物信息学软件,这些坑就可以避免啦。 例子如下:
cd ~/biosoft
mkdir samtools && cd samtools
wget https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
tar xvfj samtools-1.3.1.tar.bz2
cd samtools-1.3.1
./configure --prefix=/home/jianmingzeng/biosoft/myBin
make
make install
~/biosoft/myBin/bin/samtools --help
~/biosoft/myBin/bin/plot-bamstats --help5.2.3 系统自带软件中心
大家都知道,操作系统只是一个生态环境而已,没有上面丰富多彩的软件,它的用处很有限,就好像购买之初的手机,不下载QQ,微信,音频视频软件,根本没办法玩。同样的,做生物信息学数据分析也是如此。 唯一比较麻烦的事情是我们想安装的软件不是QQ、微信这种高频软件,而是科研相关的生物信息学数据分析软,大部分软件都不在系统自带软件中心。不过还是需要了解一下。 首先,不同的系统,安装方式不一样,windows基本没有自带软件中心,MAC有appstore,但是生物信息学相关的很少,linux根据发行版不一样,安装命令不一样,ubuntu的用apt-get,centos的用yum,其余的自己去搜索了解即可。
5.2.4 conda软件管理
正是因为软件安装的各种坑,有些软件所需环境的配置同样令人头疼,会不断报错提醒你那些东西没有安装。 而系统自带的软件中心又不太可能包含所有的软件,所以出现了conda这样的软件管理中心来弥补,详情请看[conda 官网(]https://bioconda.github.io/)
bioconda里面几乎涵盖了引用率较高的,好用的工具的打包资源,一键式安装,并且各自依赖的环境相互分隔。 每次使用source activate env_name 来激活,使用source deactivate 来退出。 具体软件列表见:https://anaconda.org/bioconda/repo 但是列表不支持搜索,可以去它的github里面去搜索 https://bioconda.github.io/
首先需要安装这个conda 在官网找到安装包:复制链接,在linux下执行如下代码:
wget https://repo.continuum.io/miniconda/Miniconda2-latest-Linux-x86_64.sh
sh Miniconda2-latest-Linux-x86_64.sh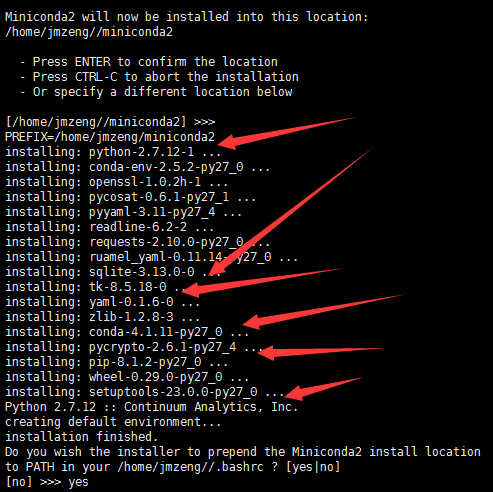 之后出现yes /no 一律选择yes,这样会默认在你的home目录下新建一个miniconda2的文件夹,然后一切默认即可~ 而且默认修改了你的环境变量文件, 即bashrc文件:输入source ~/.bashrc这样conda命令就可以使用了.
之后出现yes /no 一律选择yes,这样会默认在你的home目录下新建一个miniconda2的文件夹,然后一切默认即可~ 而且默认修改了你的环境变量文件, 即bashrc文件:输入source ~/.bashrc这样conda命令就可以使用了.
然后就可以用conda来安装其它软件,比如cutadapt,如下图所示: 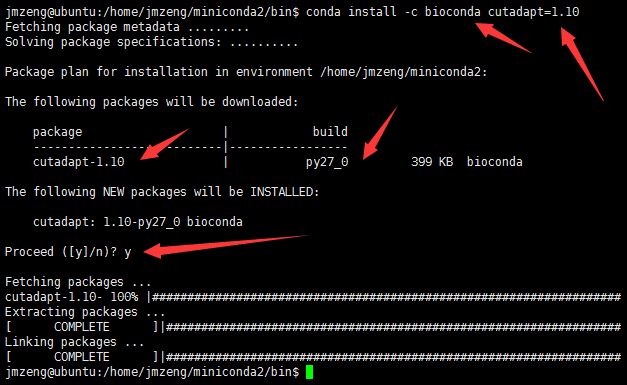 然后进入miniconda2文件夹里面的pkgs文件夹下面找到安装好的cutadapt软件,直接使用即可: python /home/jmzeng/miniconda2/pkgs/cutadapt-1.10-py27_0/bin/cutadapt –help 大功告成!!!
然后进入miniconda2文件夹里面的pkgs文件夹下面找到安装好的cutadapt软件,直接使用即可: python /home/jmzeng/miniconda2/pkgs/cutadapt-1.10-py27_0/bin/cutadapt –help 大功告成!!!
当然,也并不是所有的生物信息学相关软件都在conda的安装市场里面,如果要详细掌握它的用法,可以自己慢慢研究它的说明书,一些简单的命令如下:
conda search bwa查看可选版本
在安装时输入conda install bwa=版本号
conda list 查看所有安装的软件
conda update 软件名 可以对软件进行升级:eg. conda update bwa
conda remove 卸载已经安装的软件5.2.5 语言类软件(包)
比如perl,R,python,java,matlab,ruby,C等等
- 其中C源码就是
./configure,make,make install,也有的就是make,取决于readme,这个也是报错最多的,一般就是没有权限,缺库,很头疼。Bwa/samtools/perl/python - 然后perl和python软件呢,主要就是模块依赖的问题。Htseq/macs/circos
- R,java,软件非常简单了。Haploview/fastqc/Trimmomatic
- matlab软件,你要是在windows界面用到还好,想去linux用,也折腾好几个星期。
- ruby其它我没有用过啦。
我曾经在论坛上面发过一千个生物信息学软件安装,http://www.biotrainee.com/thread-856-1-1.html
5.3 环境变量
Linux是一个多用户的操作系统。每个用户登录系统后,都会有一个专用的运行环境。 通常每个用户默认的环境都是相同的,这个默认环境实际上就是一组环境变量的定义。 环境变量是全局的,设置好的环境变量可以被所有当前用户所运行的程序所使用。 用户可以对自己的运行环境进行定制,其方法就是修改相应的系统环境变量。
环境变量有很多,需要重点理解的就是PATH,很多时候大家看到教程某些软件的使用,比如
cd tmp/chrX_Y/hg19/
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chrX.fa.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chrY.fa.gz
gunzip chrX.fa.gz
gunzip chrY.fa.gz
~/biosoft/bwa/bwa-0.7.15/bwa index chrX.fa
~/biosoft/bwa/bwa-0.7.15/bwa mem -t 5 -M chrX.fa read*.fa >read.sam
samtools view -bS read.sam >read.bam
samtools flagstat read.bam
samtools sort -@ 5 -o read.sorted.bam read.bam
samtools view -h -F4 -q 5 read.sorted.bam |samtools view -bS|samtools rmdup - read.filter.rmdup.bam
samtools index read.filter.rmdup.bam
samtools mpileup -ugf ~/tmp/chrX_Y/hg19/chrX.fa read.filter.rmdup.bam |bcftools call -vmO z -o read.bcftools.vcf.gzbwa软件就没有添加到环境变量,所以需要用全路径,指明使用电脑里面什么地方的bwa软件来做数据分析。
而把安装好的软件添加到环境变量的方法有:
第一种方法
export PATH=/usr/local/webserver/mysql/bin:$PATH ## 先添加
echo $PATH ### 再查看上述方法的PATH 在终端关闭后就会消失。所以还是建议通过编辑/etc/profile来改PATH,也可以修改家目录下的.bashrc(即:~/.bashrc)。
第二种方法
vim /etc/profile
在最后,添加:
export PATH="/usr/local/webserver/mysql/bin:$PATH"
保存,退出,然后运行:
source /etc/profile,不报错则成功。当然,还有很多其它的环境变量,如下:
PATH: 决定了shell将到哪些目录中寻找命令或程序
ROOTPATH: 这个变量的功能和PATH相同,但它只罗列出超级用户(root)键入命令时所需检查的目录。
HOME: 当前用户主目录
USER: 查看当前的用户
LOGNAME: 查看当前用户的登录名。
UID: 当前用户的识别字,取值是由数位构成的字串。
SHELL: 是指当前用户用的是哪种Shell。
TERM : 终端的类型。
PWD 当前工作目录的绝对路径名,该变量的取值随cd命令的使用而变化。
MAIL: 是指当前用户的邮件存放目录。
HISTSIZE: 是指保存历史命令记录的条数
HOSTNAME: 是指主机的名称,许多应用程序如果要用到主机名的话,通常是从这个环境变量中来取得的。
PS1: 是基本提示符,对于root用户是#,对于普通用户是$,也可以使用一些更复杂的值。
PS2: 是附属提示符,默认是“>”。可以通过修改此环境变量来修改当前的命令符,比如下列命令会将提示符修改成字符串“Hello,My NewPrompt :) ”。# PS1=" Hello,My NewPrompt :) "
IFS: 输入域分隔符。当shell读取输入时,用来分隔单词的一组字符,它们通常是空格、制表符和换行符。5.4 编程语言
5.4.1 linux的shell
Linux系统在生物信息学数据处理中的重要性就不用我多说了,鉴于一直有学生问我一些很显而易见的问题,对系统性的学习并理解了Linux系统操作的专业人士来说是显而易见的。我在这里仅以拥有多年处理生物信息学数据经验过来人的角度给大家总结一下,Linux该如何学,该学什么,该花多少工夫,学习重点是什么?
现可以把Linux的学习过程分成三个阶段,总结如下:
第一阶段:把linux系统玩得跟windows系统一样顺畅
这一阶段的主要目的就是去可视化,熟悉黑白命令行界面。
如何远程连接服务器(使用Xshell,SecureCRT,Putty,VNC等等),了解你在服务器上面有什么权限。
左右鼠标单击双击如何实现?磁盘文件浏览如何实现?文件操作如何实现?绝对路径和相对路径区别?
需要了解的常见的Linux命令:
pwd/ls/cd/mv/rm/cp/mkdir/rmdir/man/locate/head/tail/less/more
cut/paste/join/sort/uniq/wc/cat/diff/cmp/alias
wget/ssh/scp/curl/ftp/lftp/mysql/大家可以搜每天一个linux命令的博客来跟着练习,或者看一些Linux视频(百度云盘(http://pan.baidu.com/s/1jIvwRD8 )共享了一大堆),或者关注一些Linux学习相关公众号,加入一些linux社区,论坛,当然如果你只是简单了解,搞生物信息学其实没必要那么深入理解,跟着一本像样的入门书籍(建议看鸟哥Linux私房菜),完整的学习即可!
需要深度理解的概念有:
软硬链接区别
文本编辑,文件权限设置
打包压缩解压操作(tar/gzip/bzip/ x-j x-c vf)
软件的快捷方式如何实现?
软件如何安装(源码软件,二进制可执行软件,perl/R/python/java软件)
软件版本如何管理,各种编程语言环境如何管理,模块如何管理?(尤其是大部分没有root权限)这些知识需要深度理解,所以一般初学者肯定会遇到问题,自己要多看教程和视频跟着了练习,但总会有一些不是你立即就能解决的,不要纠结,继续学习,不久之后回过头来就明白了。
翻译成生物信息学语言就是:
测序文件在哪里?测序文件有多大?测序文件的格式fastq/fasta是什么?
前几行怎么看,参考基因组如何下载?参考基因组如何建立比对索引?
blast软件如何安装以及使用?
比对结果如何看?结果如何过滤?两次结果如何比较? 建议自己安装bio-linux系统,里面会自带很多生物信息学测试数据(fastq,fasta,sam,bam,vcf,gff,gtf,bed,MAF……),安装系统的过程也是熟悉linux的过程,熟悉这些数据格式既能加强生物信息学技巧,也能练习linux操作。
第二阶段:shell脚本,类似于windows的bat批处理文件
懂很多预定义变量:.bashrc/env/HOME/
学会一些控制语句:while/if/for/ 批量执行命令
开始自定义函数,避免重复造轮子。
了解 awk/sed/grep等文件操作语言,短小精悍,很多时候可以不需要编程。
正则匹配技巧,find函数使用
了解编程技巧 ()[]{} $$ 等符合如何使用,技巧有哪些,加快你数据处理能力(建议看shell 13问) 翻译成生物信息学语言就是:
要深度组合这些命令,并且通过shell脚本,把它们在实际生物信息学数据处理中应用起来,需要很多的实践操作,可以借鉴EMBOSS软件套件,fastx-toolkit等基础软件,实现并且模仿该软件的功能。 尤其是SMS2/exonerate/里面的一些常见功能,还有DNA2.0 Bioinformatics Toolbox的一些工具。
基本上要了解到这里才能勉强算是一个合格的生物信息学工程师。
第三阶段:高级运维技巧
w/last/top/qsub/condor/apache/socket/IO/ps/who/uid/
磁盘挂载/格式化/重启系统/文件清理/IP查看/网络管理/用户管理/目录结构了解/计划任务/各种库文件了解。这个强烈建议初学者不要过于纠结,稍微了解为佳。
对于以上生信相关的Linux三个的学习阶段介绍就到这里了,牢记“不懂的名词,感觉谷歌搜索,多记笔记”。在学习Linux基础知识的同时,就可以开始项目实战,在实战的过程中要随时思考记录如何应用Linux知识辅助生物信息数据处理,并整理学习笔记以及经验分享。
其它知识点 R最新版的安装 配置ssh供远程登录 网络服务器配置lamp或者namp 虚拟机屏幕及联网设置 配置shiny,shiny-server,R-studio
5.4.2 R {# R-language}
R语言不仅在生物信息数据处理中发挥着重要作用,也是其它主流数据处理人士的首选工具。现在非常多自学生物信息学的小伙伴必须学的就是R,所以写一个R的系统性入门指导是非常有必要的。我作为老一辈的生信工程师,所以喜欢perl一点,排斥python。我也稍微看过一些python的语法,个人认为R和python几乎是一模一样的。R的特点就是内置了大量的函数,基本上你认识的英文单词都可以是一个函数,即使不是,你也可以自定义为函数。搞清楚了函数和变量,就可以看懂大部分的R代码了。
perl/python/R 的比较
python跟perl都是高级语言, 两个开发的目的不同, perl更面向过程一些,优势是严谨,快。 python主流面向对象编程, 这个跟R类似, 数据结构等方面有些不同,但可以互相调用。 实际上以上三者之间可以互相调用部分功能。python的语法并不是很严谨,个人感觉,越偏向自然语言的编程语言越通俗但不严谨,以上,是跟C比较的。 R本身起源于S语言,是主要针对统计的,也是面向对象的。本质上,是把一个比excel功能强大的软件归零化成了命令行吧。 excel高级应用也是要编程的,所以R的初级应用可以当成是没有用户交互界面的excel,细心一点, 把示例代码都打对,当功能强大但不好使版的excel吧, 这样至少心理上不会畏难跟抵触。 内部集成的越多,用户需要做的越少,你用C画个图累死你,用python得写几行,R一行就行了!
六步系统入门R语言
第一步:掌握必须要会的基础
下载R语言的软件:https://cran.r-project.org/bin/windows/base/ 下载Rstudio这个R编辑器:https://www.rstudio.com/products/rstudio/download/(在Rstudio里面写代码会比较方便)
学习help函数(你必须要把help函数用一百次以上,不然你不可能入门的!)
R的特性就是有着大量的包,所以你必须学会安装包:
安装包 install.packages(" xxxxxx ")
加载包 library( xxxxx )
查看包的帮助文档help("xxxxx") 或?xxxxx
获取当前工作区间getwd()
更改工作区间 setwd( "xxxxxx")
清除当前对象rm() 安装包你一定会遇到错误,请参考: R包终极解决方案!(http://www.biotrainee.com/thread-144-1-1.html) R的包(package)(http://www.bio-info-trainee.com/579.html)
你必须要自学R语言基础,或者看书,或者看视频,或者有人手把手教你,书的话,我推荐:《R in Action》、《The Art of_R Programming》。 这些书籍都会提供一些简单的测试代码,你跟着傻瓜式的敲代码就好. 但是实践的过程中,请务必注意一些英文单词(file文件路径/Description简述/Usage用法/Arguments参数/Details详细/value 数值/Examples例子/header 表标题/logical_value 逻辑值/delimiter 分隔符/object 对象/col列/row 行/vector向量/dimensions维度/data数据)。
第二步:明白R中的变量
向量和因子:向量特简单,没什么好说的,因子太复杂了,我说不清楚,你们慢慢理解。 数据框:就像我们的表格,第一行就是每一列的名字,我们称之为字段,或者变量名。那么对应每列下面的数据就叫做记录或者观测。用data.frame( 字段1,字段2,…. )创建 ) 列表:与数据框类似,区别就是每一列向量类型和长度可以不一致。用list( 字段1, 字段2,….. )创建 数组:其形式就像我们玩的模方,每一个面都是一个矩阵数据,用array(数据,各维度的最大值,各维度的名称)
第三步:了解变量的基础操作函数
变量怎么来,对它们处理什么?
我们处理生物信息学数据一般很少会手动创建这些对象,都是从文本里面读取,比如kegg数据库文件,差异分析结果,RNA-seq的表达量矩阵. 但是读入之后,我们的重点就是知道它们变成了什么,该如何去一步步的转换它们。 数据的特性函数也必须要知道,无非就是一些英文单词而已,你经常的玩一下,就慢慢的熟练了。 主要需要熟练的函数有:str,class,names,row.names,col.names,length,unique,view,min,max,summay,table
第四步:可视化你的变量
了解了R里面的基础变量和对象,也学会了对它们进行简单的转换,接下来就可以尝一尝R的甜头了,对任何数据都可以可视化,简简单单的就可以画一大堆的图。 plot,boxplot,barplot,pie,hist,pair,它们每个绘图函数都有自己要求的输入数据,特定的可视化结果,请务必在还没熟练使用之前help一下它们,自己主动查看它们好玩的地方,好好自学。
dev.new()新建画板
plot()绘制点线图,条形图,散点图.
barplot( ) 绘制条形图
dotchart( ) 绘制点图
pie( )绘制饼图.
pair( )绘制散点图阵
boxplot( )绘制箱线图
hist( )绘制直方图
scatterplot3D( )绘制3D散点图.
低级绘图函数:
par() 可以添加很多参数来修改图形
title( ) 添加标题
axis( ) 调整刻度
rug( ) 添加轴密度
grid( ) 添加网格线
abline( ) 添加直线
lines( ) 添加曲线
text( ) 添加标签
legend() 添加图例它们还有一系列的绘图参数(坐标轴、图例,颜色,性状,大小,空白,布局)非常繁琐,想掌握,花费的时间会非常多. 但是很多人直接跳到ggplot的绘图世界了,不想搞那么多底层绘图代码。 但是我看过一个底层R绘图集大成者,就Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes文章的作者的github里面有。但是对大部分人来说,生信的绘图,都是有套路的,其实都被别人包装成函数了,做好数据,一个函数就出了所有复杂的图。比如热图,cluster等等。
高级可视化不得不提ggplot2了,基本语法是需要学习的,但并不一定要死记硬背,最重要的是学会搜索,如下:
如何通过Google来使用ggplot2可视化 用谷歌搜索来使用ggplot2做可视化(下)
第五步:数据对象的高级操作
前面我们对向量,数据框,数组,列表都了解了,也知道如何查看数据的特性,但是要进行高级转换,就需要一些时间来学习apply系列函数,aggregate,split等函数的用法。这是一个分水岭,用好了你就算是R入门了。也可以用一些包,比如reshape2,dplyr。
当然,R里面的字符串对象是另外完全不一样的操作模式,建议大家自行搜索学习。
第六步:遨游R的bioconductor世界
这个是生物信息学特有的,也是为什么我要求搞生物信息学数据处理的人必须学习R,就是为了应用大量的bioconductor包。在这里面所有的对象都不在是基础的向量,数据框,数组,列表了,而是S3,S4对象,这个高级知识点我就不推荐了,你学会了前面的东西,就有了自己的学习经验了,后面的分分钟就搞定了。(其实你永远也搞不定的) 每学一个bioconductor的包,都是自己R水平的提升。 大家可以参考我的博客:http://www.bio-info-trainee.com/tag/bioconductor 我就是这样学习过来的。我还创建了bioconductor中国这个社区,可惜效果不好,有志者可以继续联系我,我们看看有没有可能做起来。 R语言的应用方向。
当然R肯定不只是应用在生物信息学啦,其实它在非常多的地方都有应用,尤其是金融和地理。 在如何一个方向学习R,就不仅仅是R本身的语法了,你需要学习的东西太多了。 我简单列出几个我接触过的方向吧:统计,科学计算,数据挖掘,文本挖掘,基础绘图,ggplot绘图,高级编程,都有着丰富的书籍和视频资料。 炼数成金的R七种武器系列。(强烈推荐,全套视频很容易找到)
《A Handbook of Statistical Analyses_Using_R》
《Modern Applied Statistics With S》
《Introduction to Scientific Programming and Simulation Using R》
《Mastering Scientific Computing with R》
《Practical Data Science with R》
《Data Mining explain using R》
《ggplot2 Elegant Graphics for Data Analysis》
《R Graphics Cookbook》
《R Cookbook》
《R in a Nutshell》
《R Programming for Bioinformatics》
《software for data analysis programming with R》看完以上这些,你就是R大神了。当然,前提是你看懂了也会灵活应用。
有小伙伴建议我继续以送视频送书籍的方式来增加浏览量,比如我网盘里面有几千本R语言的PDF书籍,也有十几套视频,但是,我这一篇总结写的太好了,我不想被利益被污染了,希望你可以转发给有需要的人,你的朋友会感激你的转发,让他了解这么多生信前辈的经验分享公众号!
补充:R语言学习的网络资源
R语言官方站 http://www.r-project.org/
R-blogger http://www.r-bloggers.com/
R语言资源汇总 https://github.com/qinwf/awesome-R
R语言搜索引擎 http://www.rseek.org/
R函数在线帮助 http://www.rdocumentation.org/
一个入门级的R在线教程 http://tryr.codeschool.com/
交互式的R在线教程 https://www.datacamp.com
各种cheatsheet适合打印出来随时查阅
http://cran.r-project.org/doc/contrib/Short-refcard.pdf
http://www.rstudio.com/resources/cheatsheets/
5.4.3 perl {# perl-language}
Perl是典型的脚本语言,短小精悍,非常容易上手,尤其适合处理文本,数据,以及系统管理。它在老一辈的生物信息学分析人员中非常流行,出于历史遗留原因大家肯定会或多或少地接触 Perl,即使你再怎么推崇Python或者GO等新兴编程语言。
1 入门资料
两个半小时入门指导:https://qntm.org/files/perl/perl.html 21天学完 perl,自己搜索下载PDF书籍吧! 大小骆驼书,建议都看完,以囫囵吞枣的方式阅读,只看基础知识来入门,难点全部跳过。 官网:https://www.perl.org/ 函数如何用:都可以在http://perldoc.perl.org/perl.html 查到 论坛:http://www.perlmonks.org/
2 知识要点
在看书的同时,你必须记住和熟练使用的知识点是下面这些:
理解perl里面的三种变量表示方式
$ 表示单个变量
用单双引号区别,q(),qq()
@ 表示多个变量组成的数组,qw()
% 表示关系型变量-hash
变量不严格区分类型,没有int/float/double/char这样的概念三种变量都有对应的操作技巧:
- 简单变量的操作函数: > Numerical operators: <, >, <=, >=, ==, !=, <=>, +, * > String operators: lt, gt, le, ge, eq, ne, cmp, ., x
- 数组操作(pop/push/shift/unshift/splice/map/grep/join/split/sort/reverse) # hash操作方式(keys,values,each,delete,exists)
具体需要在实战里面体会:http://www.biotrainee.com/forum-90-1.html 生信人必练的200个数据处理任务(欢迎大家去练习)
变量内容交换,字符型转为数值型,字符串转为字符数组,字符串变量,heredoc,字符串分割,字符串截取,随机数生成,取整,各种概率分布数,多维矩阵如何操作,进制转换,hash翻转,数组转hash
上下文环境
这个比较复杂: http://www.perlmonks.org/?node_id=738558,就是需要理解你写的程序是如何判断你的变量的,你以为的不一定是你以为的。
正则表达式
这也是一个非常重要的一块内容,基础用法就是m和s,一个匹配,一个替换,比较有趣的就是1,2等等捕获变量。
内建变量
就是perl语言设计的时候定义了一大堆的全局变量($_ $, $0 $> $< $! \(. @ARGV @F @_ @INC %ENV %SIG) 。外表上看起来都是一个\) @ %符号后面加上一大堆的奇奇怪怪的字符,表示一些特殊变量,这也是perl语言饱受诟病的原因。但是有些非常重要,懂了它之后写程序会方便。下载一个表格,里面有近100个预定义变量需要学习的。
控制语句(循环/条件/判断)
if … elsif … else … unless/while/next/last/for/foreach 读写文件,脚本实战!
while(<>){
#do something !
}这是我最喜欢的一个程序模板,读取文件,根据需要处理文件,然后输出。需要实现非常多的功能,然后就可以自己总结脚本技巧,也能完全掌握perl的各种语法。在生物信息学领域,需要实现的功能有!
perl 单行命令
我个人特别喜欢这个知识点,我也专门下载过一本书来学习,把这个perl单行命令教程看完就基本上能全明白。学习单行命令的前提是掌握非常多的奇奇怪怪的perl自定义变量和perl的基础语法,用熟练了之后就非常方便,很多生物信息学数据处理过程我现在基本不写脚本,都是直接写一行命令,完全代替了shell脚本里面的awk、sed/grep系列命令,就是熟悉-p -a -n -a -l -i -F -M 这些参数。
预定义函数
perl 是一个非常精简的语言,自定义的函数非常少,连min max这样常见的函数都没有,如果你需要使用这样的功能,要么自己写一个函数,要么使用加强版的包,perl的包非常多。
下面列出一些,我常用的函数:
程序必备: use/die/warn/print/open/close/<>/
数学函数:sin/cos/log/abs/rand/srand/sqrt
字符串函数 :uc/lc/scaler/index/rindex/length/pos/substr/sprintf/chop/chomp/hex/int/oct/ord/chr/unpack/unencode
defined/undef系统操作相关
perl语言是跨平台的,因为它的执行靠的是perl解释器,而perl的解释器可以安装在任何机器上面。所以可以用perl来代替很多系统管理工作。
系统命令调用
文件句柄操作(STDIN,STDOUT,STDERR,ARGV,DATA,)
系统文件管理(mkdir/chdir/opendir/closedir/readdir/telldir/rmdir/)一些高级技巧
自定义函数 sub , 参数传递,数组传递,返回值
模块操作(模块安装,加载,模块路径,模块函数引用)
引用(变量的变量)
选择一个好的编辑器-编译器,editplus,notepad++,jEdit,编程习惯的养成。
搞清楚perl版本的问题,还有程序编码的问题,中文显示的问题。 程序调试
perl常见模块学习
perl和LWP/HTML做网络爬虫必备,重点是DOM如何解析; perl和CGI编程,做网站的神器,重点是html基础知识; DBI相关数据库,用perl来操作mysql等,当然,重点是mysql知识; GD and GD::Graph 可以用来画图,但是基本上没有人用了,除了CIRCOS画圈圈图火起来了; TK模块,可以编写GUI界面程序,但是也几乎没有人用了; XML/pdf/excel/Json 相关的模块可以用来读取非文本格式数据,或者输出格式化报告; socket通信相关,高手甚至可以写出一个QQ的模仿版本; 最后不得不提的就是Bioperl了,虽然我从来没有用过,但是它的确对初学者非常有用,大多数人不提倡重复造轮子,但我个人觉得,对初学者来说,重复造轮子是一个非常好的学习方式。大家可以仿造bioperl里面的各个功能,用自己的脚本来实现!
3 复习资料
如果你感觉学的差不多了,就可以下载一些复习资料,查漏补缺: http://michaelgoerz.net/refcards/perl_refcard.pdf
https://www.cheatography.com/mishin/cheat-sheets/perl-reference-card/
http://www.catonmat.net/download/perl.predefined.variables.pdf
http://www.erudil.com/preqr.pdf
https://www.cs.tut.fi/~jkorpela/perl/regexp.html
https://support.sas.com/rnd/base/datastep/perl_regexp/regexp-tip-sheet.pdf
5.4.4 python {# python-language}
Python开发的方向太多了,有机器学习,数据挖掘,网络开发,爬虫等等。其实在生信领域,Python还显现不出绝对的优势,生信的大部分软件流程都是用shell或Perl写的,而且已经足够好用了。我选Python是因为我想顺便学点数据挖掘和机器学习的东西,而且Python这些年越来越火,发展势头远超其他脚本语言,所以学它肯定是没错的。
一、入门标准
入门比较难定义,什么程度才算入门呢?
- 掌握基本的语法,熟练使用python的内置类型、内置函数和数据结构。
- 了解一些基本的模块的使用,能够实现一些简单的需求。
后面有一个实例,如果你能简单的做完,那我敢肯定你已经入门了。
二、基本知识点
1.基本语法
缩进:Python是通过代码缩进来决定代码层次逻辑的,一般约定使用4个空格
版本问题:主要包括2.x系列的和3.x系列的,两者语法不同且不兼容,有的模块只能在指定版本下安装。建议使用3.x Python,碰到特殊问题再去使用指定版本
文件编码声明:python会去环境变量里寻找python解释器。如果代码里有中文,则要以utf-8编码
#!/usr/bin/env python
#-*- coding: utf-8 –*-变量定义:使用前要先定义
dir():列出一个数据类型或对象的所有方法,非常好用,同help()
文件操作:f = open(),f.close();with open() as f: ,os.path.exists(),os.path.isfile(),os.path.abspath()
目录操作:os.mkdir(),os.rmdir(),os.listdir(),os.chdir()
开发环境选择: - Sublime Text 对Python支持挺好,轻量级生化武器(推荐) - Eclipse+Pydev比较厚重,大型开发比较适合 - Vim/Atom - PyCharm - IPython - WingIDE
2.处理数据
2.1 基本数据类型:布尔;整型;浮点型;字符串
# 字符串的内置函数,都比较有用
'capitalize', 'casefold', 'center', 'count', 'encode', 'endswith', 'expandtabs', 'find', 'format', 'format_map', 'index', 'isalnum', 'isalpha', 'isdecimal', 'isdigit', 'isidentifier', 'islower', 'isnumeric', 'isprintable', 'isspace', 'istitle', 'isupper', 'join', 'ljust', 'lower', 'lstrip', 'maketrans', 'partition', 'replace', 'rfind', 'rindex', 'rjust', 'rpartition', 'rsplit', 'rstrip', 'split', 'splitlines', 'startswith', 'strip', 'swapcase', 'title', 'translate', 'upper', 'zfill'2.2 基本数据结构:列表、元组、字典、集合。
数据结构就是一种容器,用于在内存中存放我们的数据。
列表:任意元素组成的顺序序列,以位置为索引。
# 列表的内置函数
'append', 'clear', 'copy', 'count', 'extend', 'index', 'insert', 'pop', 'remove', 'reverse', 'sort'元组:相当于不可变的列表,防止错误修改,节省内存开销。元组解包
# 元组的内置函数
'count', 'index'字典:键值对,没有顺序,键必须是常量。
# 字典内置函数
'clear', 'copy', 'fromkeys', 'get', 'items', 'keys', 'pop', 'popitem', 'setdefault', 'update', 'values'集合:没有顺序,元素之间没有重复,相当于舍弃了值的字典。集合操作(&,|,-,^,<,<=,>,>=)
# 集合内置函数
'add', 'clear', 'copy', 'discard', 'pop', 'remove', 'update'
'isdisjoint','issuperset','issubset','symmetric_difference','difference','union', 'intersection', 'symmetric_difference_update','intersection_update','difference_update',#######2.3 控制语句 {-}
条件:if…else…
循环:for,while,break,continue
2.4 模块使用
Python有着非常友好的模块安装方法,一个pip install命令几乎可以安装绝大多数的模块。建议使用模块前多看相关API文档。
最常用的模块有:sys,os,re,csv,gzip,fileinput,random,collections,time;百度上有很多很好的模块使用入门教程。 - 正则表达式 re - 有序字典 collections.OrderedDict() - 调用系统命令 subprocess.call()
三、入门实例
题目:从大量FASTA文件中提取指定序列,并对提取到的序列做某些处理(如求反向互补序列)
描述:假设你有很多测序数据,分别存储在不同文件夹的不同文件里,现在给你一些序列名,要求你从众多数据中提取出特定的序列。
image
image
思路:遍历每一个文件夹;遍历每一个文件;读取文件,判断序列,输出序列(处理),关闭文件;处理数据,添加一个函数即可。
四、精通标准
当然这只是个噱头,精通的道路是无止境的,下面只是罗列了一些常见的高级特性。
切片,推导式,生成器,异常处理
高级模块:threading(多线程),ctypes(调用C程序优化性能),logging(日志)
专业模块:pysam - 处理基因组数据(fasta/fastq/bam/vcf)的Python模块
Biopython:Python的计算分子生物学和生物信息学工具包
编写自己的package:解决某个特定需求,上传到 PyPI,然后你就成为大神了
编程规范:写出规范化的代码 Google Python coding style
函数式编程:即使代码量暴增也不会影响代码的可读性,调试和Debug也会变得非常简单。
面向对象编程:最高级的编程方法,对函数进行分类和封装,让开发“更快更好更强…”
五、最后
Python只是一门编程语言,一种实现工具,我们可以用很多种语言来替换它,我们之所以选择Python,是因为我们喜欢它给我们带来的便捷。如果你想深入某个领域,其实真正重要的是技术背后的算法。
六、推荐资源
Python教程 - 廖雪峰的官方网站
python初级教程:入门详解
Python 面向对象(初级篇)
Python | Codecademy
Google Python编码风格
Python正则表达式指南
《Python学习手册》
《Python编程金典》
《Bioinformatics Programming Using Python》
5.4.5 R包安装终极解决方案 {# R-package}
写在前面:
我曾多次强调过R语言在生物信息学中的重要性,也激发了很多小伙伴学习的热情。 学习R语言必然会安装各种各样的包,很多人在这一步就遇到了困难。
刚开始学习R语言的时候我们经常会遇到各种包安装错误,比如package ‘airway’ is not available (for R version 3.1.0)等等,
这篇文章,我们就来系统性地整理一些新手可能遇到的问题以及解决方案。
当然,你不一定现在就会遇到,但是如果你遇到了,请记住,可以在这里得到答案!
文章目录如下:
- 查看已经安装了和可以安装哪些R包
- 如何安装旧版本的包
- 如何切换镜像以及为什么要切换
- 4种常见的R包安装方式
说明:该文首发于我的个人博客以及生信技能树论坛,请点击文末的阅读原文前往查看详细资料。
总体思路
R语言里面的包其实是很简单的,因为它自带了一个安装函数install.packages()基本上可以解决大部分问题。
但是如果出问题你需要从如下角度进行分析思考:
- 你的R语言安装在什么机器什么?(linux(ubuntu?centos?),window,mac)
- 你的R是什么版本:(3.1 ? 3.2 ? http://www.bio-info-trainee.com/1307.html )
- 你的安装器是什么版本?(主要针对于bioconductor包的安装)
- 你的联网方式是什么?https ?http ?
- 你选择的R包镜像是什么?
#####自己的R包安在哪里,可以安装哪些R包? {-}
首先在R里面输入.libPaths()即可查看当前的R把包安装到了机器的哪个地方,这样可以直接进入目录去查看有哪些包,每个包都会有一个文件夹。
其次你可以用installed.packages()查看你已经安装了哪些包。
最后你可以用available.packages()可以查看自己的机器可以安装哪些包!
>.libPaths()
[1] "C:/Users/jmzeng/Documents/R/win-library/3.1"
[2] "C:/Program Files/R/R-3.1.0/library"
colnames(installed.packages())
[1] "Package" "LibPath" "Version"
[4] "Priority" "Depends" "Imports"
[7] "LinkingTo" "Suggests" "Enhances"
[10] "License" "License_is_FOSS" "License_restricts_use"
[13] "OS_type" "MD5sum" "NeedsCompilation"
[16] "Built"
ap <- available.packages()
> dim(ap)打开ap变量可以看出,我们想安装的 airway 包根本不在,当然,这肯定是不存在的。 因为 airway 是bioconductor的包,并非R默认。
需要调整contriburl参数,如下:
> dim(available.packages(contriburl = "https://cran.rstudio.com/bin/windows/contrib/3.2/"))
[1] 8110 17
> dim(ap)
[1] 8155 17
> dim(available.packages(contriburl = "http://bioconductor.org/packages/3.1/bioc/bin/windows/contrib/3.2/"))
[1] 1000 17
> dim(available.packages(contriburl = "http://mirrors.ustc.edu.cn/bioc//packages/3.1/bioc/bin/windows/contrib/3.2/"))
[1] 1000 17用这个参数,可以看不同仓库,甚至不同版本的R包共有哪些资源!
如何安装旧版本的包?
既然你点进来看,肯定是有需求。 一般来说,R语言自带的
install.packages函数来安装一个包时,都是默认安装最新版的。 但是有些R包的开发者他会引用其它的一些R包,但是它用的是旧版本的功能,自己来不及更新或者疏忽了。 而我们又不得不用他的包,这时候就不得不卸载最新版包,转而安装旧版本包。
首先你要用remove.packages这个命令把现在的包卸载掉!
然后去包的官网上面找到它的旧版本的下载链接:
我这里拿ggplot2举例: http://cran.r-project.org/src/contrib/Archive/ggplot2/
#packageurl <- "http://cran.r-project.org/src/contrib/Archive/ggplot2/ggplot2_1.0.1.tar.gz"
install.packages(packageurl, repos=NULL, type="source")
#我这里安装它的1.0.1版本,而不是最新版!
#还有很多其它方法,我就不一一举例了,这个是我认为最方便,最直观的!
# install yesterday's version of checkpoint, by date
install.dates('checkpoint', Sys.Date() - 1)
# install earlier versions of checkpoint and devtools
install.versions(c('checkpoint', 'devtools'), c('0.3.3', '1.6.1'))很明显,我是在*StackOverflow**上面搜索得到的解决方案,O(∩_∩)O哈哈~ 你可以参考:http://stackoverflow.com/questions/17082341/installing-older-version-of-r-package
如何切换镜像
这个技巧很重要,一般来说,R语言自带的install.packages函数来安装一个包时,都是用的默认的镜像!
如果你是用的Rstudio这个IDE,默认镜像就是:https://cran.rstudio.com/
如果你直接用的R语言,那么就是:http://cran.us.r-project.org
但是一般你安装的时候会提醒你选择,而我们需要更改成自己最方便的
install.packages(pkgs, lib, repos = getOption("repos"),
contriburl = contrib.url(repos, type),
method, available = NULL, destdir = NULL,
dependencies = NA, type = getOption("pkgType"),
configure.args = getOption("configure.args"),
configure.vars = getOption("configure.vars"),
clean = FALSE, Ncpus = getOption("Ncpus", 1L),
verbose = getOption("verbose"),
libs_only = FALSE, INSTALL_opts, quiet = FALSE,
keep_outputs = FALSE, ...)如果是在国内,install.packages("ABC",repos="http://mirror.bjtu.edu.cn/ "),换成北大的镜像你会体验飞一般的感觉!
如果想永久设置,就用options修改即可。
如果你是Rstudio的IDE,只需要鼠标点击直接进入全局设置,一劳永逸的选择好镜像!
插图
你可以check一下每个镜像的包是不是一致的:
dim(available.packages(contriburl = "http://cran.rstudio.com/bin/windows/contrib/3.2/"))更改镜像主页及包的版本即可查看所有镜像各提供哪些包!
当然,我们的bioconductor其实也是有镜像的,只是大部分人都不知道,也不会去用而已!
source("http://bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
biocLite("RGalaxy")
##这样就用中科大的镜像来下载包啦
##bioconductor还有很多其它镜像:https://www.bioconductor.org/about/mirrors/
##https://stat.ethz.ch/R-manual/R-devel/library/utils/html/chooseBioCmirror.html4种常见的R包安装方式
R自带函数直接安装
这个是最简单的,而且不需要考虑各种包之间的依赖关系。
对普通的R包,直接install.packages()即可,一般下载不了都是包的名字打错了,或者是R的版本不够。如果下载了安装不了,一般是依赖包没弄好,或者你的电脑缺少一些库文件,如果实在是找不到或者下载慢,一般就用repos=来切换一些镜像。
> install.packages("ape") ## 直接输入包名字即可
Installing package into ‘C:/Users/jmzeng/Documents/R/win-library/3.1’
(as ‘lib’ is unspecified) ##一般不指定lib,除非你明确知道你的lib是在哪里
trying URL 'http://mirror.bjtu.edu.cn/cran/bin/windows/contrib/3.1/ape_3.4.zip'
Content type 'application/zip' length 1418322 bytes (1.4 Mb)
opened URL ##根据你选择的镜像,程序会自动拼接好下载链接url
downloaded 1.4 Mb
package ‘ape’ successfully unpacked and MD5 sums checked
##表明你已经安装好包啦
The downloaded binary packages are in
##程序自动下载的原始文件一般放在临时目录,会自动删除
C:\Users\jmzeng\AppData\Local\Temp\Rtmpy0OivY\downloaded_packages对于bioconductor的包,我们一般是
source("http://bioconductor.org/biocLite.R") ##安装BiocInstaller
#options(BioC_mirror=”http://mirrors.ustc.edu.cn/bioc/“) 如果需要切换镜像
biocLite("ggbio")
#或者直接
BiocInstaller::biocLite('ggbio')
## 前提是你已经安装好了BiocInstaller
#某些时候你还需要卸载
remove.packages("BiocInstaller")
#然后安装新的进入主页找到包下载地址
可以选择用R自带的下载器来下载,也可以把下面的url拷贝到浏览器用浏览器来下载
packageurl <- "http://cran.r-project.org/src/contrib/Archive/ggplot2/ggplot2_0.9.1.tar.gz"
packageurl <- "http://cran.r-project.org/src/contrib/Archive/gridExtra/gridExtra_0.9.1.tar.gz"
install.packages(packageurl, repos=NULL, type="source")
#packageurl <- "http://www.bioconductor.org/packages/2.11/bioc/src/contrib/ggbio_1.6.6.tar.gz"
#packageurl <- "http://cran.r-project.org/src/contrib/Archive/ggplot2/ggplot2_1.0.1.tar.gz"
install.packages(packageurl, repos=NULL, type="source")这样安装的就不需要选择镜像了,也跨越了安装器的版本!
下载到本地后再安装
download.file("http://bioconductor.org/packages/release/bioc/src/contrib/BiocInstaller_1.20.1.tar.gz","BiocInstaller_1.20.1.tar.gz")
##也可以选择用浏览器下载这个包
install.packages("BiocInstaller_1.20.1.tar.gz", repos = NULL)如果你用的RStudio这样的IDE,那么直接用鼠标就可以操作了。或者用choose.files()来手动选择把下载的源码BiocInstaller_1.20.1.tar.gz放到哪里。但这种形式大部分安装都无法成功,因为R包之间的依赖性很强!
命令行版本安装
如果是linux版本,命令行从网上自动下载包如下:
sudo su - -c \
"R -e \"install.packages('shiny', repos='https://cran.rstudio.com/')\""如果是linux,命令行安装本地包,在shell的终端
sudo R CMD INSTALL package.tar.gzwindow或者mac平台一般不推荐命令行格式,可视化那么舒心,何必自讨苦吃呢?
5.4.6 perl模块终极解决方案 {# perl-module}
这种细节问题问我,我当然无法直接给出答案咯。毕竟,我的知识积累都不是靠死记硬背的。所以需要取回过头查看一下我的博客,才意识到,
我在博客陆陆续续写了7篇教程,关于perl的模块。目录如下:
- ubuntu服务器解决方案第七讲-perl安装模块
- Perl用cpan在linux上面安装模块
- Perl及R及python模块碎碎念
- perl模块终极解决方案-上
- perl模块终极解决方案-下
- perl程序技巧-检验系统环境或模块安装
首先需要自己确定已经安装了哪些模块,都安装在哪里?还有新的模块需要安装到哪里? 然后再学习如何安装新的模块。
一、装Perl模块有两种方法
- 自动安装 (使用CPAN模块自动完成下载、编译、安装的全过程)
- 手工安装 (去CPAN网站下载所需要的模块,手工编译、安装)
二、使用CPAN模块自动安装
安装前需要先联上网,有无root权限均可。
初次运行CPAN时需要做一些设置,运行下面的命令即可:
perl -MCPAN -e shell如果你的机器是直接与因特网相联(拨号上网、专线,etc.),那么一路回车就行了,只需要在最后一步选一个离您最近的 CPAN 镜像站点。例如我选的是位于国内的http://www.cnblogs.com/itech/admin/ftp://www.perl87.cn/CPAN/
如果你的机器位于防火墙之后,还需要设置ftp代理或http代理。
其实大部分人的机器都不需要走这一步的,肯定是用过了perl的cpan功能啦,除非你是新买的电脑。
下面是常用 cpan 命令。
cpan>help
cpan>m
cpan>install Net::Server
cpan>quit我简单解释一下吧:
查询:cpan[1]> d /模块名字或者部分名字/
查询结果中会给出所有含有模块名字或者部分名字的模块,选择您所需要的模块进行下载
下载安装:cpan[1]> install 模块名字
同时会自动安装很多依赖的模块,非常方便。三、手工安装的步骤
一般情况下不推荐这种安装方式,但是总是会有迫不得已的时候,而且尝试这种方式,能加深对perl模块的理解。
比如从 CPAN下载了Net-Server模块0.97版的压缩文件Net-Server-0.97.tar.gz,假设放在/usr/local/src/下。
cd /usr/local/src
tar xvzf Net-Server-0.97.tar.gz
cd Net-Server-0.97
perl Makefile.PL
make test如果测试结果报告all test ok,你就可以放心地安装编译好的模块了。 安装模块前,先要确保你对你下载包的文件夹(例子里面是/usr/local/src/)有可写权限(通常以 su 命令获得). 当然,只有root用户才会/usr/local/src/有写入的权限,普通用户把模块文件下载到自己的文件夹即可。
测试自己的模块安装成功与否,用下面的命令,如果没有给出任何输出,那就没问题。
perl -MNet::Server -e1上述步骤适合于 Linux/Unix下绝大多数的Perl模块。可能还有少数模块的安装方法略有差别,所以最好先看看安装目录里的 README 或 INSTALL。
有的时候如果是build.pl的需要以下安装步骤:(需要Module::Build模块支持)
perl Build.PL
./Build
./Build test
./Build install四、cpan和root权限的关系
前面我说过,是否有root权限,都可以调用cpan下载器的,但还是有些微区别的。
如果是root用户,模块其实没有问题,直接用cpan下载器,几乎能解决所有的模块下载安装问题!
但是如果是非root用户,那么就麻烦了,很难用自动的cpan下载器,总有一些模块用cpan下载失败。
这样只能下载模块源码,然后编译,但是编译有个问题,很多模块居然是依赖于其它模块的,你的不停地下载其它依赖模块,最后才能解决,特别麻烦! 但是我仍然不推荐大家用手工下载的方式安装perl模块。 这里我推荐所有的非root用户运行下面的代码获取自己的私人cpan下载器。
wget -O- http://cpanmin.us | perl - -l ~/perl5 App::cpanminus local::lib
eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`
echo 'eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`' >> ~/.profile
echo 'export MANPATH=$HOME/perl5/man:$MANPATH' >> ~/.profile就能拥有一个私人的cpan下载器,~/.profile可能需要更改为.bash_profile, .bashrc, etc等等,取决于你的linux系统! 然后你直接运行cpanm Module::Name,就跟root用户一样的可以下载模块啦! 或者用下面的方式在shell里面安装模块,其中ext是模块的安装目录,可以修改!
perl -MTime::HiRes -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Time::HiRes;
perl -MFile::Path -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Path;
perl -MFile::Basename -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Basename;
perl -MFile::Copy -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Copy;
perl -MIO::Handle -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext IO::Handle;
perl -MYAML::XS -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML::XS;
perl -MYAML -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML;
perl -MXML::Simple -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext XML::Simple;
perl -MStorable -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Storable;
perl -MStatistics::Descriptive -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Statistics::Descriptive;
perl -MTie::IxHash -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Tie::IxHash;
perl -MAlgorithm::Combinatorics -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Algorithm::Combinatorics;
perl -MDevel::Size -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Devel::Size;
perl -MSort::Key::Radix -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key::Radix;
perl -MSort::Key -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key;
perl -MBit::Vector -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Bit::Vector;
perl -M"feature 'switch'" -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext feature;五、非root用户的另一个解决方案
手动下载local::lib, 这个perl模块,然后自己安装在指定目录,也是能解决模块的问题!
下载之后解压,进入:
perl Makefile.PL --bootstrap=~/.perl ##这里设置你想把模块放置的目录
make test && make install
echo 'eval $(perl -I$HOME/.perl/lib/perl5 -Mlocal::lib=$HOME/.perl)' >> ~/.bashrc等待几个小时即可!!!
添加好环境变量之后,就可以用
perl -MCPAN -Mlocal::lib -e 'CPAN::install(LWP)'这样的模式下载模块了,所有的模块都会存储在$HOME/.perl/lib/perl5 里面!!! 如果是新写的perl程序,需要在开头加入 use local::lib;
这样才能sets up a local lib at ~/perl5,才能使用该模块!
当然每次写程序添加这个也实在是太麻烦了,其实你也可以直接打开 ~/.bashrc,然后写入下面的内容
PERL5LIB=$PERL5LIB:/PATH_WHERE_YOU_PUT_THE_PACKAGE/source/bin/perl_module;
#(笨蛋,这个里面的内容-路径-是需要你修改的,别直接拷贝粘贴哈)
export PERL5LIB可以把perl模块安装在任何地方,然后通过这种方式去把模块加载到你的perl程序!
PATH="/home/jmzeng/perl5/bin${PATH:+:${PATH}}"; export PATH;
PERL5LIB="/home/jmzeng/perl5/lib/perl5${PERL5LIB:+:${PERL5LIB}}"; export PERL5LIB;
PERL_LOCAL_LIB_ROOT="/home/jmzeng/perl5${PERL_LOCAL_LIB_ROOT:+:${PERL_LOCAL_LIB_ROOT}}"; export PERL_LOCAL_LIB_ROOT;
PERL_MB_OPT="--install_base \"/home/jmzeng/perl5\""; export PERL_MB_OPT;
PERL_MM_OPT="INSTALL_BASE=/home/jmzeng/perl5"; export PERL_MM_OPT;
六、查看perl模块的安装目录
这里指的是查看那些被添加到了环境变量的perl模块安装目录,理论上你可以在如何文件夹里面安装一个perl模块,但是如果不添加到环境变量,意义不大,因为大多数perl程序只会在环境变量里面搜索安装的perl模块,其它地方的模块它们无法调用。
主要就是@INC这个默认变量 ,可以用下面的代码查看:
perl -e '{print "$_\n" foreach @INC}'比如我其中一个服务器显示如下:
/home/jmzeng/perl5/lib/perl5/5.18.2/x86_64-linux-gnu-thread-multi
/home/jmzeng/perl5/lib/perl5/5.18.2
/home/jmzeng/perl5/lib/perl5/x86_64-linux-gnu-thread-multi
/home/jmzeng/perl5/lib/perl5
/etc/perl
/usr/local/lib/perl/5.18.2
/usr/local/share/perl/5.18.2
/usr/lib/perl5
/usr/share/perl5
/usr/lib/perl/5.18
/usr/share/perl/5.18
/home/jmzeng/perl5/lib/perl5/5.18.1
/usr/local/lib/site_perl七、查看已经安装哪些perl模块
不管你有没有root权限,进入 cpan 然后install ExtUtils::Installed模块 这样就可以执行 instmodsh 这个命令了,可以查看当前环境下所有的模块! 为什么可以直接使用呢,因为模块安装的时候就顺便把instmodsh给你添加到了环境变量,你可以用 which instmodsh 查看它被安装到哪里了。
/usr/bin/instmodsh
/home/jmzeng/perl5/bin/instmodsh当然也可以写出脚本来利用这个模块查询其它模块安装信息,主要是写脚本校验用户电脑模块的时候用得着。
#!/usr/bin/perl
use strict;
use ExtUtils::Installed;
my $inst= ExtUtils::Installed->new();
my @modules = $inst->modules();
foreach(@modules)
{
my $ver = $inst->version($_) || "???";
printf("%-12s -- %s\n", $_, $ver);
}
exit 0; 八、模块理论上可以安装到如何地方
比如非root用户,使用 cpan ,那么一般会创建/home/yourname/.cpan这个隐藏目录下面存储个人的perl模块。 因为不是root用户,所以cpan并不是万能的,有些包是安装不成功的,比如GD模块 而且也可以直接下载模块文件,自己编译到任何目录,只需要在运行自己的脚本的时候加上下面一句话。
use lib '/home/your-home/perl_lib';但是,大部分情况下,我们安装模块不是因为我们自己写脚本需要,而且一些生物信息学软件对模块有依赖,但是我们很少有能力修改那些生物信息学软件。 所以这条路一般是不走的。 如果有很多自己下载的包,统一安装到了一个目录,就可以把该目录添加目录到@INC。
5.4.7 Python包安装的小结 {# python-package}
想当初刚学习Python的时候,就会用书本里面自带的一些package,用sys,os也用得很开心。 后来接触到biopython项目,发现原来Python有这么不同功能的包,简直琳琅满目。 不过这也是我痛苦的开始,在服务器上装个包怎么那么费劲呢,缺这少那的。 为了一个包的安装,我得花多少时间啊,还能不能让人好好做科研了。
一、黑暗时代
最开始的时候是从源码开始安装,一般python setup.py install就执行安装过程了,不过可怕的这些包之间的依赖关系。而且安装的时候,要选择安装目录。对于刚开始学习的我,都要搞晕了。后来还看到一个叫easy_install,可以自动解析package之间的依赖关系,生产效率感觉提上去了。不过经常出错,虽然比手动安装好多了,使用起来还是挺费劲的。
二、迎来曙光
不知道当时从哪里看到说用pip会更好,看来没事上上网还是挺有好处的。 而且比easy_install什么的不知道高到哪里去了,具体差异可以看pip vs easy_install和why use pip over easy_install,上面的链接都说得很详细了。对于一般的需求,pip install --user <package>就已经很受用了。如果再使用上豆瓣上的PyPi源,那使用体验简直不能太好。
# Linux/Mac用户修改
# $HOME/.config/pip/pip.conf
[global]
timeout = 60
index-url = https://pypi.doubanio.com/simple
## 注意: 如果使用http链接,需要指定trusted-host参数
[global]
timeout = 60
index-url = http://pypi.douban.com/simple
trusted-host = pypi.douban.com常用的pip用法一般有:
# 在用户目录安装软件,不需要root 权限
pip install --user <package>
# 搜索package
pip search biopython
# 安装特定版本的package,版本号可以从search的结果中找到
pip install biopython=1.69
# 卸载package
pip uninstall biopython
# 导出已安装的包信息
pip freeze > requirements.txt
# 其他使用方法可以参考pip的帮助说明
pip -h三、发现virtualenv
如果你只是测试,或者电脑上同一个package安装了好几个版本,那么你一定会喜欢virtualenv。 有了它,现在可以在电脑上安装不同版本的package了。 使用方法也很简单,因为virtualenv也是Python包,可以直接用pip来进行安装。 现在可以用它在电脑上创建不同的虚拟环境了,各个虚拟环境互不干扰,而且对原有的环境不会造成影响,哪天不想玩了,直接把对应的目录删掉就可以了,非常方便 。
# 安装virtualenv
pip install --user virtualenv
# 创建一个新的环境
mkdir my_envs
cd my_envs
# 创建一个env_test目录,把相关的package安装到该目录下
virtualenv env_test
# 如果系统上有多个python版本,可以通过参数来指定对应的python版本
virtualenv -p /usr/bin/python2.7 env2.7
# 激活虚拟环境,需要提供具体的虚拟环境安装目录
source env2.7/bin/activate
# 激活后就可以在终端中看到有对应提示,如果想关闭也很简单
deactivate上面的都一些基本的用法,如果创建的虚拟环境比较多,可以借助virtualenvwrapper来进行管理,更多的信息可以参考Python虚拟环境。
四、大杀器anaconda
当时在学习virtualenv的时候,也发现anaconda这个东西,不过当时觉得软件太多,而且比较臃肿。 对我这种没装几个软件的来说,virtualenv已经够用了。直到有一天我需要安装tensorflow的时候,才发现这东西有多方便。 不仅帮你解决依赖关系,而且还会帮你把相关的系统依赖也解决了。相信大家在安装软件的时候,没少遇到missing 什么 libxxxx.so什么的信息。 有的时候为了安装这些系统依赖,真的能把人搞疯。anaconda不仅能解决这些问题,还可以安装R里面package啊, 虽然我没用过这个功能。 现在除了anaconda,还有精简的miniconda和专门为生物信息准备的bioconda,虽然名字不一样,只是默认安装时带的package不一样而已,使用方法没什么区别。 下面简单说明下bioconda的使用:
# 首先需要安装conda,我们下载minicoda,文件比较小,下载过程比较快
wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh这样在你的$HOME目录里面有一个miniconda,保存安装的软件使用,而且自动在环境变量配置文件.bashrc添加新的变量设置。 安装之后可以自己检查一下,是不是有新的不一样的东西。你需要要重新登录一下或者重新加载环境变量source ~/.bashrc。 因为网络环境问题,最好修改一下安装软件源,可以使用清华anaconda开源镜像源
# 请注意一上顺序哈
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels defaults
conda config --add channels r
conda config --add channels bioconda# 搜索特定软件包
conda search package-name
# 新建一个叫py3的环境,这个环境里面我们需要指定使用python3
conda create -n py3 python=3.5.3
# 激活虚拟环境
source activate py3
# 关闭虚拟环境
source deactivate
# 列出已经创建的虚拟环境
conda info --evns五、参考