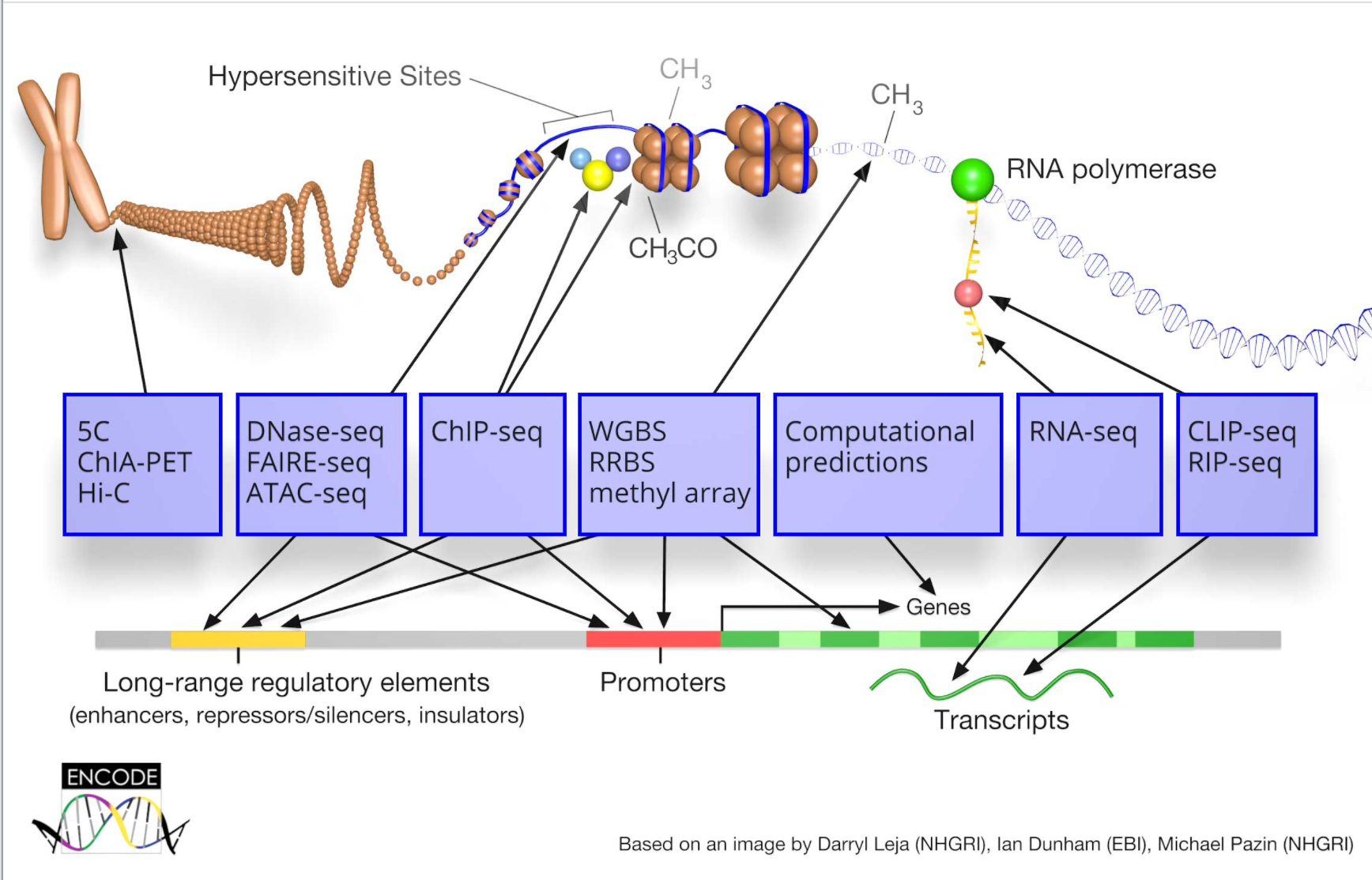
3 生物信息学数据库资源
做数据分析常常会需要用到参考基因组和注释文件,还会需要分析公共数据,了解常见的生物信息学数据库资源也是非常有必要的!
3.1 基因ID
到目前为止,仅仅是人类研究,就有两万五左右的蛋白编码基因,这些基因可以合成十几万种蛋白质,还有近十万的编码lncRNA的基因,近万的miRNA等非编码基因。基因在生物信息学研究中具有中心地位,所以对于基因的命名也显得至关重要。
每个领域,每个地域,都有权威的科研单位,他们偏向于自己定义各种各样的基因命名系统,并没有一个统一的命名方式。
而且为了研究基因,还有产生探针捕获的技术,各个厂商的探针ID也是五花八门。
在疾病研究领域,也需要独特的ID。
一些功能数据库也会对自己的样本,基因重新编码ID。
常见的基础数据库也会提出自己的ID (entrez ID, Symbol, RefSeq, probeset, PubmedID,OminID,Accnum),甚至,你自己整理发表一个数据库也可以提出基因命名系统,当然,不一定会那么受欢迎,也不会有那么多人去学习你的命名规则。
为什么要有这么多的基因ID呢?基因就像每个人一样,都是独特的个体。它在不同的地点扮演不同的角色。故自然有不同的ID,有的根据它的位置区分。比如:王总。有的根据它的特征区分比如:吝啬王。这样就会出现很多的称呼,即ID.,但他说的都是同一个人哦。
基因也是一样的,当在表达谱数据的时候,他的名字就是探针,当在ENSEMBL中时,就是ENSG开头加数字的格式,所以在不同的数据库中会有不同的命名故就会有很多的基因ID.
ID种类繁多
其中GeneCards数据库里面列出了128种数据库ID,虽然很全面,但并非都是重点,希望大家把学习时间花在刀刃上,有一些就不要死记硬背了。 而且,我觉得大部分人看到了下面这些密密麻麻的ID,肯定是要疯掉的。 一般初学者常见的ID转换工具就是DAVID了,但其实可以自己用R编程的各种包来做转换,这样自己知道自己在做什么,也了解ID是如何定义的。
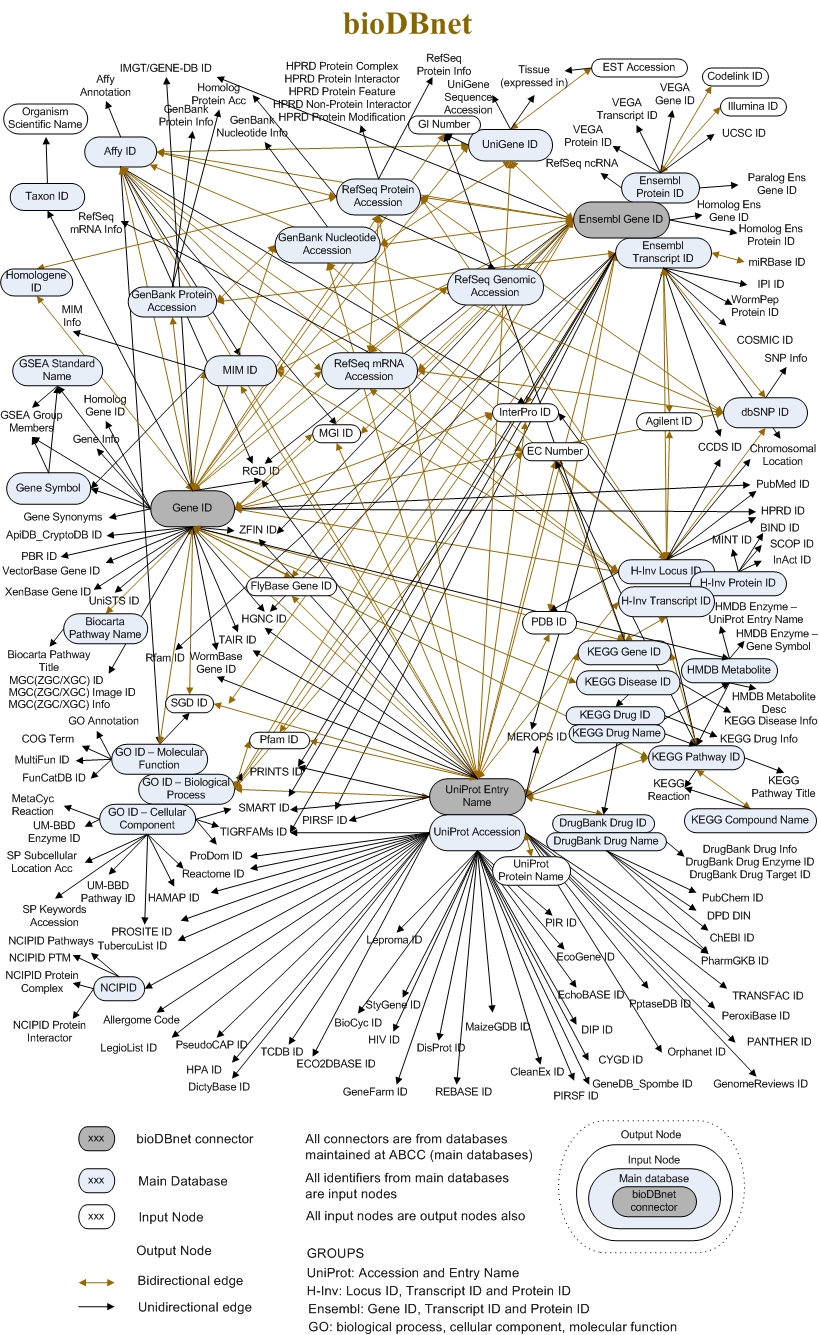
常见的数据库ID
虽然有综合性的数据库收集整理了百余种ID,使得它们之间的对应及转换得以顺利进行,但大部分数据分析过程中并不会用到所有的数据ID,最常见的如下表。
| ID 示例 | ID 来源 |
|---|---|
| ENSG00000116717 | Ensemble ID |
| GA45A_HUMAN | UniProtKB/Swiss-Prot, entry name |
| A5PJB2_BOVIN | UniProtKB/TrEMBL, entry name |
| A2BC19, P12345, A0A022YWF9 | UniProt, accession number |
| GLA, GLB, UGT1A1 | HGNC Gene Symbol |
| U12345, AF123456 | GenBank, NCBI, accession number |
| NT_123456, NM_123456, NP_123456 | RefSeq, NCBI, accession number |
| 10598, 717 | Entrez ID, NCBI |
| uc001ett, uc031tla.1 | UCSC ID |
Ensembl stable ID 的结构是根据不同物种设置的前缀, 加上数据所指的类型, 如基因蛋白质, 再加上一系列的数字. 有的时候可以有不同的版本, 则在 Ensembl ID 后面加上小数点和版本号.
常用物种前缀
| 前缀 | 学名 |
|---|---|
| ENSCEL | Caenorhabditis elegans (Caenorhabditis elegans) |
| ENSCAF | Canis lupus familiaris (Dog) |
| ENSDAR | Danio rerio (Zebrafish) |
| FB | Drosophila melanogaster (Fruitfly) |
| ENS | Homo sapiens (Human) |
| ENSMUS | Mus musculus (Mouse) |
| ENSRNO | Rattus norvegicus (Rat) |
| ENSXET | Xenopus tropicalis (Xenopus) |
类型前缀
| 前缀 | 类型 |
|---|---|
| E | exon |
| FM | Ensembl protein family |
| G | gene |
| GT | gene tree |
| P | protein |
| R | regulatory feature |
| T | transcript |
UniProt 中录入的数据都被分配了一个唯一的 entry name,叫做UniProtKB/Swiss-Prot entry name。它是最多有 11 位包含大写字母的字符串, 一般有着 “X_Y” 的形式, 其中 “X” 是最多五个便于记忆的蛋白质编号, “_” 是下划线, “Y” 是最多五个便于记忆的物种编号.
蛋白质编号示例如下
| Code(X) | Recommended protein name | Gene name |
|---|---|---|
| B2MG | Beta-2-microglobulin | B2M |
| HBA | Hemoglobin subunit alpha | HBA1 |
| INS | Insulin | INS |
| CAD17 | Cadherin-17 | CDH17 |
物种编号示例如下
| Code | Species |
|---|---|
| BOVIN | Bovine |
| CHICK | Chicken |
| ECOLI | Escherichia coli |
| HORSE | Horse |
| HUMAN | Homo sapiens |
| MAIZE | Maize (Zea mays) |
| MOUSE | Mouse |
| PEA | Garden pea (Pisum sativum) |
| PIG | Pig |
| RABIT | Rabbit |
| RAT | Rat |
| SHEEP | Sheep |
| SOYBN | Soybean (Glycine max) |
| TOBAC | Common tobacco (Nicotina tabacum) |
| WHEAT | Wheat (Triticum aestivum) |
| YEAST | Baker’s yeast (Saccharomyces cerevisiae) |
| HUGO | Gene Nomenclature Committee |
Gene Symbol
Gene Symbol 是用来表示基因的编码, 由大写字母构成, 或由大写字母和数字构成, 首字母均应该是字母,有点像是是基因的标准缩写。
如:
- GLA这个symbol代表着”galactosidase, alpha”
- GLB这个代表着”galactosidase, beta”
- UGT1A1这个symbol代表着”UDP glycosyltransferase 1 family, polypeptide A1”
GenBank Accession Number
GenBank 的通用 accession number 通常是由一个大写字母加上 5 个数字的组合, 或者两个大写字母加上 6 个数字的组合.
RefSeq Accession Number
RefSeq 有一套特殊的 Accesion Number. 形式是: [A-Z]{2}[_][0-9]{6:}, 两个大写字母, 一个下划线, 6 个或更多的数字.
Accession
| 前缀 | 类型 | 说明 |
|---|---|---|
| AC_ | Genomic | Complete genomic molecule, usually alternate assembly |
| NC_ | Genomic | Complete genomic molecule, usually reference assembly |
| NG_ | Genomi c | Incomplete genomic region |
| NT_ | Genomic | Contig or scaffold, clone-based or WGS |
| NW_ | Genomic | Contig or scaffold, primarily WGS |
| NS_ | Genomic | Environmental sequence |
| NZ_ | Genomic | Unfinished WGS |
| NM_ | mRNA | |
| NR_ | RNA | |
| XM_ | mRNA | Predicted model |
| XR_ | RNA | Predicted model |
| AP_ | Protein | Annotated on AC_ alternate assembly |
| NP_ | Protein | Associated with an NM_ or NC_ accession |
| YP_ | Protein | |
| XP_ | Protein | Predicted model, associated with an XM_ accession |
| ZP_ | Protein | Predicted model, annotated on NZ_ genomic records |
Entrez ID
Entrez 是 NCBI 使用的能够对众多数据库进行联合搜索的搜索引擎, 其对不同的 Gene 进行了编号, 每个 gene 的编号就是 entrez gene id. 由于 entrez id 相对稳定, 所以也被众多其他数据库, 如 KEGG 等采用. Entrez Gene ID 就是一系列数字, 也比较容易辨识. R 或网站都有众多的工具可以帮助从不同的 ID 转换为 entrez id 或者反向转换. 生信菜鸟团的博客《NCBI的基因entrezID相关文件介绍》讲解了Entrez ID主要的信息文件。可以直接搜索。 一个简单的Entrezid对应的别的ID,例子如下:
| gene_id | symbol | chromosome |
|---|---|---|
| 352937 | dio2 | 20 |
上表中geneid即为 Entrezid. 在ID转换中有重要的作用。
UCSC ID
由小写字母和数字构成, 起始均为 uc, 然后是三位数字, 接着又是三位小写字母, 最后有小数点和数字构成版本号. 如: uc010qfk.3, uc010qfk.3. 这个ID几乎被抛弃不用了,只是因为UCSC是三大数据库之一而已。
ID 转换
最重要的就是怎么实现ID转换。常用的id有entrez gene ID, HUGO symbol, refseq ID, ensembl ID。他们之间进行转换,做一些后续的分析。一般初学者用的ID转换的工具就是DAVID,R里面关于ID转换常用的包为org.Hs.eg.db这一类的包。 在生信技能树论坛里,健明发的《ID转换大全》和《生信人必须了解的各种ID表示方式》以及《生信编程直播第8题-几个ID转换咯》里面有实战的代码,务必运行一遍。在论坛直接搜索即可。
方法1:直接在DAVID网站,粘贴转换
方法2:用R包。不管是什么ID转换,都是找到对应关系,然后match一下即可!–《生信编程直播第8题-几个ID转换咯》有完整代码
方法3:用R包,基于org.Xx.eg.db系列包,进行ID转换。–《ID转换大全》由完整代码
总之:
entrez gene ID 文盲不会写汉字,只能运用纯数字
Ensembl ID 有文化,身前物种做玉坠
refseq ID最懒惰,一躺在中间,字母在两边
Gene Symbol大写字母加数字,一生平庸最常见直达车:
http://www.biotrainee.com/thread-941-1-1.html
http://www.biotrainee.com/thread-862-1-1.html
3.2 参考基因组版本
不同版本对应关系
hg19,GRCH37和ensembl75是三种国际生物信息学数据库资源收集存储单位,即NCBI,UCSC和ENSEMBL各自发布的基因组信息。
hg系列,hg18/19/38来自UCSC也是目前使用频率最高的基因组。从出道至今我就只看过hg19了,但是建议大家都转为hg38,因为它是目前的最新版本。
基因组各种版本对应关系综合来看如下所示:
- GRCh36 (hg18): ENSEMBL release_52.
- GRCh37 (hg19): ENSEMBL release_59/61/64/68/69/75.
- GRCh38 (hg38): ENSEMBL release_76/77/78/80/81/82.
ENSEMBL的版本特别复杂也很容易搞混,UCSC的版本就简单很多,常用的是hg19,最新版本为hg38。
看起来NCBI也是很简单,就GRCh36,37,38,但是里面水也很深!
Feb 13 2014 00:00 Directory April_14_2003
Apr 06 2006 00:00 Directory BUILD.33
Apr 06 2006 00:00 Directory BUILD.34.1
Apr 06 2006 00:00 Directory BUILD.34.2
Apr 06 2006 00:00 Directory BUILD.34.3
Apr 06 2006 00:00 Directory BUILD.35.1
Aug 03 2009 00:00 Directory BUILD.36.1
Aug 03 2009 00:00 Directory BUILD.36.2
Sep 04 2012 00:00 Directory BUILD.36.3
Jun 30 2011 00:00 Directory BUILD.37.1
Sep 07 2011 00:00 Directory BUILD.37.2
Dec 12 2012 00:00 Directory BUILD.37.3从上面可以看到,有37.1, 37.2和 37.3 等等,不过这种版本一般指的是注释在更新而基因组序列一般不变。
总之你需要记住, hg19基因组大小是3G,压缩后八九百兆。
如果要下载GTF注释文件,基因组版本尤为重要。
NCBI:最新版(hg38)
- ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/GFF/
NCBI:其它版本
- ftp://ftp.ncbi.nlm.nih.gov/genomes/Homo_sapiens/ARCHIVE/
Ensembl
- ftp://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.gtf.gz
变化上面链接中的release就可以拿到所有版本信息
- ftp://ftp.ensembl.org/pub/
UCSC
它本身需要一系列参数:
1. Navigate to http://genome.ucsc.edu/cgi-bin/hgTables
2. Select the following options:
clade: Mammal
genome: Human
assembly: Feb. 2009 (GRCh37/hg19)
group: Genes and Gene Predictions
track: UCSC Genes
table: knownGene
region: Select "genome" for the entire genome.
output format: GTF - gene transfer format
output file: enter a file name to save your results to a file, or leave blank to display results in the browser
3. Click 'get output'.搞清楚版本关系了,接下来就是进行下载。UCSC里面下载非常方便,只需要根据基因组简称来拼接url:
http://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/mm9/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/chromFa.tar.gz或者用shell脚本指定下载的染色体号
for i in $(seq 1 22) X Y M;
do echo $i;
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;done
gunzip *.gz
for i in $(seq 1 22) X Y M;
do cat chr${i}.fa >> hg19.fasta;
done
rm -fr chr*.fasta3.3 NCBI
NCBI (National Center for Biotechnology Information,美国国立生物技术信息中心)于1988年11月4日建立,是NIH(美国国立卫生研究院)的NLM(国立医学图书馆)的一个分支。目的是通过提供在线生物学数据和生物信息学分析工具来帮助人类更好的认知生物学问题。 目前有将近40个在线的文库和分子生物学数据库,包括:PubMed, PubMed Central, and GenBank等。网址: https://www.ncbi.nlm.nih.gov/
一、任务
- 为储存和分析分子生物学、生物化学、遗传学知识创建自动化系统;
- 从事研究基于计算机的信息处理过程的高级方法,用于分析生物学上重要的分子和化合物的结构与功能;
- 促进生物学研究人员和医护人员应用数据库和软件;
- 努力协作以获取世界范围内的生物技术信息。
二、内容
1.文献数据库
包括:PubMed,PubMed Central,Books等
2.序列资源库
包括人,小鼠,果蝇,线虫等各种物种的基因组数据库
包含DNA,RNA,蛋白等各种类型的数据
如:SNP,GEO,SRA等
3.常用序列分析工具
Entrez – 数据挖掘的工文本条件查询工具(Text Term Searching) 来自于超过10万个种物的核酸和蛋白序列数据,连同蛋白三维结构,基因组图谱信息和文献信息检索 网址:https://www.ncbi.nlm.nih.gov/gquery/
BLAST – 序列比对工具
https://blast.ncbi.nlm.nih.gov/Blast.cgi
4.数据下载与上传
数据下载接口:ftp://ftp.ncbi.nlm.nih.gov/
上传的工具有:Sequin,tbl2asn等,链接地址:https://www.ncbi.nlm.nih.gov/guide/data-software/
5.其他合作项目
我们比较常用的就是检索文献,检索序列,比对序列。了解更多内容可以参考官网手册:https://www.ncbi.nlm.nih.gov/books/NBK143764/
参考资料
https://baike.baidu.com/item/NCBI/3598184?fr=aladdin
https://www.ncbi.nlm.nih.gov/books/NBK143764/
3.3.1 GEO
基因表达数据库(GEO,Gene Expression Omnibus database,https://www.ncbi.nlm.nih.gov/geo/ )是由NCBI负责维护的一个数据库,设计初衷是为了收集整理各种表达芯片数据,但是后来也加入了甲基化芯片,lncRNA,miRNA,CNV芯片等各种芯片,甚至高通量测序数据,是目前最大、最全面的公共基因表达数据资源。所有的数据均可以在ftp站点下载:ftp://ftp-trace.ncbi.nih.gov/geo/
首先,我们在GEO的主页( https://www.ncbi.nlm.nih.gov/geo/ )可以看到:
Browse Content
Repository Browser
DataSets: 4348
Series: 87717
Platforms: 17572
Samples: 2165066截止到2017年8月17日,统计信息如上,可以看到数据量已经很恐怖了。
一、GEO数据库基础知识
GEO Dataset (GDS) 数据集的ID号
GEO Series (GSE) study的ID号
GEO Platform (GPL) 芯片平台
GEO Sample (GSM) 样本ID号这些数据都可以在ftp里面直接下载。
二、数据上传
上传的方式:
- 网页
- Excel表格
- 软件
- MINiML格式上传
详细上传方法,参见:https://www.ncbi.nlm.nih.gov/geo/info/submission.html
提交Affymetrix芯片数据到GEO数据库 http://www.biotrainee.com/thread-810-1-1.html
三、数据挖掘
- Entrez GEO-DataSets
官网: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gds
收录整个实验数据,可以通过技术类型,作者,物种和实验变量等信息来进行搜索。一旦相关数据被查询到,可以通过提供上面的小工具做一些分析,比如:热电图分析,表达分析,亚群的影响等
2.Entrez GEO-Profiles
官网:https://www.ncbi.nlm.nih.gov/geoprofiles/
收录单个基因的表达谱数据。可以通过基因名字,GenBank编号,SAGE标签,GEO编号等来进行搜索
3.GEO BLAST
GEO Blast界面容许用户根据核酸序列的相似性来搜索相关的GEO-Profiles 所有的BLAST结果中“E”的标签代表这个数据跟GEO-Profiles表达数据相关。
- 数据下载
我们一般是拿到了GSE的study ID号,然后直接把什么的url修改一下,就可以看到关于该study的所以描述信息,是用的什么测序平台(芯片数据,或者高通量测序),测了多少个样本,来自于哪篇文章! 所有需要的数据均可以下载,而且都是在上面的ftp里面可以根据规律去找到的,甚至可以自己拼接下载的url链接,来做批量化处理!
例如:用GSE75528,则在https://www.ncbi.nlm.nih.gov/geo/ 官网上直接搜索GSE75528 或直接输入 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75528 修改这个url最末尾的GSE号码就可以进入自己想去的任何研究的GEO页面。
如果是芯片数据,那么就需要自己仔细看GPL平台里面关于每个探针对应的注释信息,才能利用好别人的数据。 如果是高通量测序数据,一般要同步进入该GSE对应的SRA里面去下载sra数据,然后转为fastq格式数据,自己做处理!
四、其他
- 联系方式
上传数据或查询数据有问题,可以联系 geo@ncbi.nlm.nih.gov
- 写一个Python脚本下载GEO数据
脚本逻辑很简单:
- 根据GEO accession找到FTP地址
- 用wget循环下载FTP地址下的数据
#!/bin/python3
import refrom urllib.request
import urlopen
import os
def main(geo):
# find the FTP address from [url=https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GEO]https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GEO[/url]
response = urlopen("https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc={}".format(geo))
pattern = re.compile("<a href=\"(.*?)\">\(ftp\)</a>")
# use wget from shell to download SRA data
ftp_address = re.search(pattern,response.read().decode('utf-8')).group(1)
os.system(' wget -nd -r 1 -A *.sra ' + ftp_address)
if __name__ == '__main__':
from sys import argv
main(argv[1])- 数据分析
5行代码搞定GEO学习总结版:http://www.biotrainee.com/thread-612-1-1.html
参考资料:
http://www.bio-info-trainee.com/1835.html
https://www.ncbi.nlm.nih.gov/geo/info/GEOHandoutFinal.pdf
3.3.2 SRA
跟GEO类似,NCBI的SRA(Sequence ReadArchive,https://www.ncbi.nlm.nih.gov/sra/ )数据库是专门用于存储二代测序的原始数据,包括 454, IonTorrent, Illumina, SOLiD, Helicos and CompleteGenomics等。 除了原始序列数据外,SRA现在也存在raw reads在参考基因的aligment information。
该数据库也是International Nucleotide Sequence Database Collaboration (INSDC) 的一部分。INSDC包含:NCBI Sequence Read Archive (SRA), European Bioinformatics Institute (EBI), 和 DNA Database of Japan (DDBJ)。数据提交给其中任何一个数据库中后,数据都是共享的。
一、数据库结构
每个数据库都有自己最小的可发表单元。例如:PubMed最小可发表单元是一篇文献,SRA中最小可发表单元是一次实验(标签为:SRX#)。
NCBI中SRA数据结构的层次关系:Studies,Experiments, Samples,Runs:
Studies是就实验目标而言的,一个study可能包含多个experiment。
Experiments包含了样本,DNA source,测序平台,数据处理等信息。
一个experiment可能包含一个或多个runs。
Runs 表示测序仪运行所产生的reads.SRA数据库用不同的前缀加以区分:ERP or SRP for Studies, SRS for samples, SRX for Experiments, and SRR for Runs。
二、数据上传
登陆NCBI账号
注册你的项目和生物样本
注册项目:https://www.ncbi.nlm.nih.gov/bioproject/
注册样本:https://www.ncbi.nlm.nih.gov/biosample/
上传SRA数据
上传SRA metadata (关于该项目、实验的等信息)
上传序列数据
更详细的说明,参见 https://www.ncbi.nlm.nih.gov/sra/docs/submit/
三、数据下载
如果要下载每个study对应的runs的所有数据,我们需要下载安装SRA Toolkit!
链接地址: http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
SRA toolkit常用命令的说明文档见:
http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc。
这里我们需要使用prefetch命令进行下载
$prefetch SRR776503 SRR776505 SRR776506下载完成后,会在你的工作主目录下生成一个ncbi的文件夹。
Sra子文件夹中的.sra文件就是对应的runs文件。 ’.sra’的后缀是SRA数据库对fastq文件的特殊压缩。使用前,我们需要将其解压为fastq文件。SRA Toolkit 包含了解压函数fastq-dump :$fastq-dump SRR776503.sra
通过命令行来下载
for ((i=204;i<=209;i++)) ;
do
wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP017/SRP017311/SRR620$i/SRR620$i.sra;
done
ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 $id;done四、其他
- 上传或下载数据遇到问题,可咨询:
sra@ncbi.nlm.nih.gov
参考资料
http://www.biotrainee.com/thread-800-1-1.html
https://www.ncbi.nlm.nih.gov/sra/docs/
3.3.3 dbSNP
单核苷酸多态性(single nucleotide polymorphism,SNP) 主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。它是人类可遗传变异中最常见的一种,占所有已知多态性的90%以上。dbSNP(The Single Nucleotide Polymorphism database) 是一个公共的核酸多态性的数据库,它是关于单碱基替换以及短插入、删除多态性的资源库。网址:https://www.ncbi.nlm.nih.gov/projects/SNP。
一、最新版本
人的dbsnp目前已更新到150版本 150版本基本信息
| Organism | dbSNP Build | Genome Build | Number of Submissions(ss#’s) | Number of RefSNP Clusters (rs#’s) ( # validated) | Number of (rs#’s) in gene | Number of (ss#’s) with genotype | Number of (ss#’s) with frequency |
|---|---|---|---|---|---|---|---|
| Homo sapiens | 150 | 38.3 | 907,234,193 | 325,660,549 (107,926,145) | 191,665,918 | 73,917,935 | 130,169,906 |
下载地址:ftp://ftp.ncbi.nlm.nih.gov/snp/
二、目录结构
点击:ftp://ftp.ncbi.nlm.nih.gov/snp/ 则进入snp网址。
dbSNP包含了许多目录,其中最有用的是:
organisms/
database/
specs/相关详细信息见 ftp://ftp.ncbi.nih.gov/snp/00readme.txt
FTP的“specs/”目录包含重要的文件的格式,内容及其基本介绍。
FTP的“organisms/”目录包含了一列有SNP数据的生物体目录,其按通用名后接NCBI分类id号来组织的。(要知道,DBsnp不光包含人的数据哦,还有bacteria,tuna等物种的snp信息;人类物种ID是9606,可以看到variation位点信息有基于hg19和hg38的两种下载方式,如果还有其它需求,可以自己用基因组坐标转换工具。)
点击特定的生物体子目录即可访问其中的ftp报表文件,你还可以选择同一个物种的不同版本。比如你点击了human_9606目录,那么你会发现人类数据目录包含以下子目录:ASN1_bin/、ASN1_flat/、XML/、VCF/、chr_rpts/、gene_report/、Genome_report/、rs_fasta/、ss_fasta/、genotype_by_gene/、genotype/、haplotypes/、database/、misc/、Enterz/
- ASN1_bin : ASN.1 二进制格式的RefSNP文档综述 (.bin)
- ASN1_flat :从ASN.1 二进制格式而来的按染色体排序的RefSNP docsum(.flat)
- XML: 提供refSNP簇的具体查询信息以及NCBI SNP交换格式的簇成员(.xml)
- chr_rpts :包含特定染色体上的RefSNPs 的完整列表(.txt)
- genotype :以基因型交换XML格式提供提交的SNPs的submitter和基因型信息(.xml)
- genome_reports :包含生物体SNP密度分布的概要报告以及每个基因中的SNPs的概要报告(无后缀或.rpt或.log)
- ss_fasta :包含FASTA格式的生物体的所有可用的submitted SNP(ss)序列数据(.fas)
- rs_fasta :包含FASTA格式的人类所有可用的参考SNP (RS)序列数据(.fas)
chr_rpts 中的txt文件内容:
RefSNP id (rs#)rs代号
mapweight where 匹配个数
- 1 = Unmapped
- 2 = Mapped to single position in genome
- 3 = Mapped to 2 positions on a single chromosome
- 4 = Mapped to 3-10 positions in genome (possible paralog hits)
- 5 = Mapped to >10 positions in genome.
snp_type where snp类型
Total number of chromosomes hit by this RefSNP during mapping 匹配到的染色体个数
Total number of contigs hit by this RefSNP during mapping 匹配到的conting个数
Total number of hits to genome by this RefSNP during mapping 匹配到基因组的个数
Chromosome for this hit to genome 匹配到的染色体
Contig accession for this hit to genome 匹配到conting 序号
Position of RefSNP in contig coordinates 在conting中匹配到突变的位置
Position of RefSNP in chromosome coordinates (used to order report)
在染色体中匹配到突变的位置
* x, a single number, indicates a feature at base position x
* x..y, denotes a feature that spans from x to y inclusive.
* x^y, denotes a feature that is inserted between bases x and yGenes at this same position on the chromosome 匹配到的基因名字
Genotypes available in dbSNP for this RefSNP 基因型是否可知
- 1 = yes
- 0 = no
bed中包含各个染色体上的snp,如下:
chrom: The name of the chromosome (e.g. chr1, chr2, etc.).
chromStart: The Reference SNP (rs) start position on the chromosome.
Note: The first base in a chromosome is numbered 0.
- chromEnd: The rs end position on the chromosome.
Optional Fields:
name: The dbSNP Reference SNP (rs) ID
score: dbSNP does not assign a score value, so this field will always contain a 0 .
strand: This field defines strand orientation as either + or -.
VCF 这个是dbSNP数据库的精髓文件,需要仔细理解,内容节选如下:
#CHROM POS ID REF ALT QUAL FILTER INFO
1 948136 rs267598747 G A . . RS=267598747;RSPOS=948136;dbSNPBuildID=137;SSR=0;SAO=3;VP=0x050060000305000002100120;GENEINFO=NOC2L:26155;WGT=1;VC=SNV;PM;REF;SYN;ASP;LSD;CLNALLE=1;CLNHGVS=NC_000001.11:g.948136G>A;CLNSRC=.;CLNORIGIN=2;CLNSRCID=.;CLNSIG=1;CLNDSDB=MedGen:SNOMED_CT;CLNDSDBID=C0025202:2092003;CLNDBN=Malignant_melanoma;CLNREVSTAT=no_assertion_provided;CLNACC=RCV000064926.2它包含的内容:染色体,突变的位置,rs代号,突变过程,info。
其中info包含了突变是否为同义突变?突变实在coding 区还是内含子或UTR?也包含了clinvar数据库的临床意义信息,CLNSIG(0 - Uncertain significance, 1 - not provided, 2 - Benign, 3 - Likely benign, 4 - Likely pathogenic, 5 - Pathogenic, 6 - drug response, 7 - histocompatibility, 255 - other);CLNDSDB(Variant disease database name);CLNDBN(Variant disease name)还有更多解释,直接看第二章的VCF格式介绍即可。
三、查询
http://www.ncbi.nlm.nih.gov/SNP/ 是NCBI做好的一个网页版查询工具,因为下载一个 variation位点信息记录文件动辄就是十几个G,一般人也不会处理那个文件,不知道从里面应该如何提取需要的信息,这时候学习它的网页版查询工具也挺好的。
在UCSC里面也有对dbsnp数据库的介绍,主要是从数据库设计的角度来理解,里面详细介绍了每一列具体的意义,值得大家仔细学习。
http://genome.ucsc.edu/cgi-bin/hgTables?db=hg19&hgta_group=varRep&hgta_track=snp146&hgta_table=snp146&hgta_doSchema=describe+table+schema
http://genome.ucsc.edu/cgi-bin/hgTables?db=hg19&hgta_group=varRep&hgta_track=snp141&hgta_table=snp141&hgta_doSchema=describe+table+schema
但是如果真想从数据库语言的角度来理解,需要看它的数据库设计的schema了:很复杂:ftp://ftp.ncbi.nih.gov/snp/database/erd_dbSNP.pdf
sql的代码也可以下载: ftp://ftp.ncbi.nlm.nih.gov/snp/organisms/human_9606/database/organism_schema/
还根据gene来分genotype:ftp://ftp.ncbi.nlm.nih.gov/snp/organisms/human_9606/genotype_by_gene/
四、命名
关于snp位点的命名其实并不统一,大家在文献中一般用的都是习惯或者说惯用名称。这里只介绍NCBI的rs号。NCBI里对所有提交的snp进行分类考证之后,都会给出一个rs号,也可称作参考snp,并给出snp的具体信息,包括前后序列,位置信息,分布频率等,应该说用这个rs号是比较容易确定搞明白的。 一般写法是这样: dbSNP后面跟featureID。featureID一般是rs/ss后跟7-8位数字, 比如: rs12345678或者dbSNP|rs12345678 。
最后值得一提的是,除了dbsnp对variation规定了ID号,还有几个其它偏门的ID号也可以来描述变异位点的。
NCBI的dbsnp,以rs和ss开头
illumina的kgp开头
ESP的以esp开头
kgp是illumina中华八芯片的五、其他
有任何疑问可联系:snp-admin@ncbi.nlm.nih.gov
参考资料:
http://www.bio-info-trainee.com/1863.html
http://blog.sina.com.cn/s/blog_751bd9440102w6rm.html
https://www.ncbi.nlm.nih.gov/books/NBK21088/
3.3.4 RefSeq
NCBI RefSeq (Reference Sequence,美国国立生物技术信息中心参考序列库) 是目前世界上最具有权威性的序列数据库。NCBI的参考序列计划(RefSeq)将为中心法则中自然存在的分子,从染色体到mRNA到蛋白提供参考序列标准。RefSeq标准为人类基因组的功能注解提供一个基础。它们为突变分析,基因表达研究,和多态发现提供一个稳定的参考点。
- 全面的,整合的,无冗余的序列
- 基因组DNA,RNA,蛋白产物
- 是医学、功能、多样性研究的一个基准
- 为基因组注释,基因鉴定和特性描述,突变和多态性分析,表达研究和比较分析提供稳定可靠的参考
- 由NCBI和其合作者维护
| Proteins | Transcripts | Organisms |
|---|---|---|
| 88,385,530 | 19,634,664 | 71,356 |
– 最新数据截止2017年7月21日
由于一些序列来自异常连接产生的转录物或由计算机推演产生的不正确内含子-外显子剪切,因此该数据库所收集的参考序列一直在不断地被修改中,尽管如此,NCBI RefSeq 仍是目前最可信赖的人类基因mRNA序列数据库。
一、命名
RefSeq一般的命名格式:前缀为两个字母,然后下横线(’_’)。区别于其它的GenBank的命名格式。
Model RefSeq: XM_ (mRNA), XR_ (non-coding RNA), and XP_ (protein) 这个是首先被提交的
Known RefSeq: NM_ (mRNA), NR_ (non-coding RNA), or NP_ (protein) 代表被人工检验过- 在Comment区域显示来源,说明数据可靠性。(GENOME ANNOTATION,INFERRED,MODEL, PREDICTED,PROVISIONAL,WGS REVIEWED,VALIDATED)
- 蛋白序列在DBSOURCE区域标示 ‘REFSEQ’
blast结果中序列名的含义
blast一般返回的结果序列开头的格式都如正下面所示:
gi|4557284|ref|NM_000646.1|[4557284]格式说明:
- gi :”GenBank Identifier的缩写”, 是序列的ID号,标识符。唯一的。
- 4557284 就是该序列的gi号
- ref :标示该序列是参考序列。
- NM_000646.1 该序列的Accession号和版本号
预测的,临时的,和检查过的RefSeq记录有什么区别?
RefSeq记录是有三种可以获得的状态:预测的,临时的和检查过的(reviewd)。
检查过的RefSeq记录代表了目前关于一个基因和它的转录子的知识的汇编。它们很多都来自于GenBank记录、人类基因组命名委员会、和OMIM。RefSeq标准为人类基因组的功能注解提供一个基础。
预测的RefSeq记录是来自于那些未知功能的cDNA序列,它们有一个预测的蛋白编码区。
临时的RefSeq记录还没有被检查过。它们是有自动的程序产生的。
二、如何访问RefSeq
- BLAST
http://blast.ncbi.nlm.nih.gov/blast/
将序列跟已经注释的序列比对,寻找序列之间的差异
- Clinical Remap
www.ncbi.nlm.nih.gov/genome/tools/remap
比较重新组织的序列跟RefSeqGene序列之间的差异
- Variation Reporter
http://www.ncbi.nlm.nih.gov/variation/tools/reporter/
报到突变跟RefSeq序列的关系
- 其他会检索RefSeq库的工具
mapview https://www.ncbi.nlm.nih.gov/mapview/
ENTREZ GENE https://www.ncbi.nlm.nih.gov/gene
ENTREZ GENOMES DIVISION https://www.ncbi.nlm.nih.gov/genome
- 数据下载
下载地址:ftp://ftp.ncbi.nlm.nih.gov/refseq/
其它物种: ftp://ftp.ncbi.nlm.nih.gov/refseq/release/
三、讨论
- RefSeq和genbank的数据有什么区别?
genbank是一个开放的数据库,对每个基因都含有许多序列。很多研究者或者公司都可以自己提交序列,另外这个数据库每天都要和EMBL和DDBJ交换数据。genbank的数据可能重复或者不准。 而RefSeq数据库被设计成每个人类位点挑出一个代表序列来减少重复,是NCBI提供的校正的序列数据和相关的信息。数据库包括构建的基因组contig、mRNA、蛋白和整个染色体。refseq序列是NCBI筛选过的非冗余数据库,一般可信度比较高。
- 为什么RefSeq记录中的基因符号(symbol)有时和相关的GenBank中的不一样?
RefSeq全部使用官方基因符号。而GenBank是一个公共的序列备份库,由数据发现者提供。有的作者会向相关的物种命名委员会取得官方基因符号,但有的作者没有,所以有时会产生别名。GenBank与Pubmed相同,通过display可以选择显示格式,常用的有GenBank和FASTA两种格式。如果要对基因序列作进一步分析,FASTA格式是很好的选择。FASTA格式仅包括该序列的简要特征,并以ATGC4种碱基列出核苷酸序列,简单明了。而GenBank格式可显示较完整的基因序列记录,反映核苷酸序列的详细信息
参考资料
http://www.ncbi.nlm.nih.gov/refseq/
http://liucheng.name/379/
http://yangl.net/2015/10/08/ncbi_refseq/
http://yangl.net/2015/10/08/ncbi-refseq-name-format/
http://www.biotrainee.com/thread-213-1-1.html
https://www.ncbi.nlm.nih.gov/books/NBK21091/
ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/RefSeqGene/presentations/RefSeqGene.pptx
3.3.5 Entrez
Entrez (http://www.ncbi.nlm.nih.gov/Entrez) 是美国国家生物技术信息中心所提供的在线资源检索器。该资源将GenBank序列与其原始文献出处链接在一起。 Entrez是由NCBI主持的一个数据库检索系统。
一、Entrez系统数据库
有将近38个库,这里仅列举了部分,具体请参见( https://www.ncbi.nlm.nih.gov/gquery/gquery.fcgi )
- Literature
- Books: 在线生物医学图书
- PubMed 生物医学文献数据库
- Health
- OMIM : 人类孟德尔遗传数据库
- Genomes
- SRA:二代测序的原始数据
- SNP: 单核苷酸多肽性数据库
- Taxonomy: GenBank 中的物种分类学数据库
- Genes
- GEO: 基因表达数据库
- Proteins
- Structure: 大分子三维结构数据库
- Structure: 大分子三维结构数据库
- Chemicals
- BioSystems 跟基因,蛋白,化学分子关联的分子通路
二、Entrez检索的方法
- 检索规则
- 词间默认逻辑关系为AND
- 短语检索加引号“”;
- 使用的逻辑运算符有AND、OR 和NOT, 但必须大写;
- 支持截词检索, 截词符用*表示;
- 定义词条类型:[ ]
- 用:表示起始
例子:
从左到右的顺序,关联词大写
promoters OR response elements NOT human AND mammals
先执行括号里面的逻辑
g1p3 AND (response element OR promoter)
horse[Organism]
neoplasms[MeSH Terms]
prolactin[Protein Name]
srcdb_refseq[Properties]
2010/06[Publication Date]
110:500[Sequence Length]
2015/3/1:2016/4/30[Publication Date]
PubMed: ("horses"[MeSH Terms] OR "horses"[All Fields] OR "horse"
[All Fields] OR "equidae"[MeSH Terms] OR "equidae"[All Fields])
AND ("receptors, dopamine d2"[MeSH Terms] OR ("receptors"[All Fields]
AND "dopamine"[All Fields] AND "d2"[All Fields]) OR "dopamine d2
receptors"[All Fields] OR ("dopamine"[All Fields] AND "receptor"
[All Fields] AND "d2"[All Fields]) OR "dopamine receptor d2"[All Fields])
Protein: ("Equus caballus"[Organism] OR horse[All Fields]) AND (dopamine
receptor D2[Protein Name] OR (dopamine[All Fields] AND receptor[All Fields]
AND D2[All Fields])
模糊匹配
NC_0000*[Accession] AND Human[Organism]2.搜索
- 图形界面的搜索
在主页 https://www.ncbi.nlm.nih.gov/ 选择好数据库,进行检索。
NCBI上所有的资源见:https://www.ncbi.nlm.nih.gov/guide/all/
登陆NCBI以后会保留你的搜索记录。
进入单独的数据库搜索界面,会有advanced选项,更精细的搜索:
Nucleotide: www.ncbi.nlm.nih.gov/nucleotide
PubMed: www.ncbi.nlm.nih.gov/pubmed
Gene: www.ncbi.nlm.nih.gov/gene/advanced- 直接输入网址
蛋白编号gi4557757,GenPept格式(默认)
www.ncbi.nlm.nih.gov/protein/4557757
核酸编号,NM_000240和NM_000041,GenBank格式
www.ncbi.nlm.nih.gov/nucleotide/NM_000240,NM_000041&report=genbank
Gene编号348
www.ncbi.nlm.nih.gov/gene/348
Gene编号348,XML格式
www.ncbi.nlm.nih.gov/gene/348?report=XML
PubMed ID为9705509和19745054,abstract格式
www.ncbi.nlm.nih.gov/pubmed/9705509,19745054?report=abstract&format=text
在nucleotide中搜索APOE基因,限制一页呈现200个结果
www.ncbi.nlm.nih.gov/nucleotide/?term=APOE[gene]&dispmax=200
在PubMed中搜索Lipman DJ和PMID的格式呈现 www.ncbi.nlm.nih.gov/pubmed/?term=Lipman+DJ&report=uilist
- 命令行的搜索
可以通过E-utilities(Entrez Programming Utilities )来进行批量的下载或检索。
感兴趣的可以参考:https://www.ncbi.nlm.nih.gov/books/NBK25501/
bioython也带有相关的工具:http://biopython-cn.readthedocs.io/zh_CN/latest/cn/chr09.html
参考资料
《NCBI的Entrez系统检索技巧》
https://www.ncbi.nlm.nih.gov/books/NBK3837/
3.4 Ensembl
Ensembl是由EBI和Sanger共同开发的真核生物基因组注释项目,它侧重于脊椎动物的基因组数据,但也包含了其他生物如线虫,酵母,拟南芥和水稻等,其中,BioMart是用户提取Ensembl基因组数据的强大工具。
Ensembl项目得到的数据均可以通过其基因组浏览器查看,用于支持脊椎动物基因组的比较基因组,进化,序列突变和转录调控方面研究。Ensembl注释基因,多重序列比对,预测结构和收集疾病数据。Ensembl工具包括:BLAST, BLAT, BioMart 和 Variant Effect Predictor (VEP)。
一、简介
Ensembl是由英国Sanger研究所Wellcome基金会(WTSI)和欧洲分子生物学实验室所属分部欧洲生物信息学研究所(EMBI-EBI)共同协作运营的一个项目。这些机构均位于英国剑桥市南部辛克斯顿的威康信托基因组校园(Wellcome Trust Genome Campus)内。
Ensembl计划开始于1999年,人类基因组草图计划完成前的几年。即使在早期阶段,也可明显看出,三十亿个碱基对的人工注释是不能够为科研人员提供实时最新数据的获取的。因此Ensembl的目标是自动的基因组注释,并把这些注释与其他有用的生物数据整合起来,通过网络公开给所有人使用。Ensembl数据库网站开始于July 2000,是一个真核生物基因组注释项目,其侧重于脊椎动物的基因组数据,但也包含了其他生物,如线虫,酵母,拟南芥和水稻等。近年来,随着时间推移,越来越多的基因组数据已经被添加到了Ensembl,同时Ensembl可用数据的范围也扩展到了比较基因组学、变异,以及调控数据。
目前Ensembl的组员有40到50个人,分成几个小组:
Genebuild小组负责不同物种的gene sets创建。他们的结果被保存在核心数据库中,该数据库由Software小组进行运维。Software小组还负责BioMart数据挖掘工具的开发和维护。
Compara、Variation以及Regulation小组分别负责比较组学、突变以及调控的数据相关工作。
Web小组的工作是确保所有的数据能够在网站页面上,通过清晰和友好用户界面呈现出来。
Production小组负责Ensembl数据的常规更行。
最后,Outreach小组负责用户的答疑,以及提供全球范围内使用Ensembl的研讨会议或知识培训。
截止到2017年7月,Ensembl发发布了最新的Ensembl 90版本数据
包含的基因组的物种:http://asia.ensembl.org/info/about/species.html
基因注释的数据来源
- 最新的基因组数据(大部分是动物)
- UniProt/Swiss-Prot和UniProt/TrEMBL蛋白序列
- NCBI RefSeq蛋白和核酸序列
- EMBL cDNA序列
二、Ensembl可以做什么
- 查看基因在染色体上的注释
- 查看基因的选择性转录
- 探索某个基因的超过50个物种的同源性和进化树
- 比较物种的全基因组的比对和保守区域
- 查看比对到Ensembl上的芯片序列
- 查看染色体任何一区域的ESTs, clones, mRNA和proteins
- 检查染色体或基因上的SNPs (single nucleotide polymorphisms)
- 查看不同品种(rat,mouse),种群,品种(狗)的SNPs
- 查看比对到Ensembl基因上的mRNA或蛋白的序列位置
- 上传自己的数据
- 通过BLAST或BLAT来搜索Ensembl基因组中相似的序列
- 通过BioMart导出序列和基因信息
- Variant Effect Predictor
三、下载
- 少量的数据
大多数Ensembl 基因组数据的描述页有”export”功能,可以直接导出这一页的内容。
- 大的数据集
PERL API http://www.ensembl.org/info/docs/api/index.html
如果不熟悉Perl语言,可以通过Ensembl REST API http://rest.ensembl.org/
- 复杂的交叉数据库
BioMart http://www.ensembl.org/info/data/biomart/index.html
- 全部的数据集
FTP site http://www.ensembl.org/info/data/ftp/index.html
四、其他
- Ensembl genes命名
人的基因
ENSG Gene
ENST Transcript
ENSE Exon
ENSP Protein
例如: ENST00000252723其他物种的基因,例如老鼠(Mus musculus)
ENSMUSG Mouse Gene
ENSMUST Mouse Transcript
ENSMUSE Mouse Exon
ENSMUSP Mouse Protein- 常见问题
http://www.ensembl.org/Help/Faq
参考资料
http://asia.ensembl.org/info/about/index.html
http://www.ensembl.org/info/index.html
3.5 UCSC
下面我们来介绍一下作为生信人必须掌握的三大数据库 NCBI-UCSC-ENSEMBL之一的UCSC。
一、简介
2000年6月22日,UCSC(University of California,Santa Cruz)和其他国际人类基因组计划的成员完成了人基因组组装的第一个草图,并承诺永久对外提供基因组信息。几个星期以后,在2000年7月22日,组装的基因组在网站 ttp://genome.ucsc.edu 呈现出来,并提供了一个在线的查询分析工具UCSC Genome Browser。接下来的几年里,该网站不断的发展,如今已包含大量的脊椎动物和模式生物的基因组组装和注释信息,并停工了一系列查看,分析,下载数据的工具。
站点地址:
- http://genome.ucsc.edu/
- Europe: http://genome-euro.ucsc.edu
- Asia: http://genome-asia.ucsc.edu
数据库特点:
- 给浏览基因组数据提供了可靠和迅速的方式。
- 整合了大量的基因组注释数据,约有一半的注释信息是UCSC通过来自公开的序列数据计算得出,另外一半来自世界各地的科学工作者。本身并不下任何结论,而只是收集各种相关信息供用 户参考。
- 支持数据库检索和序列相似性搜索。
二、UCSC可以干什么
UCSC建立的初衷是为了更好的呈现基因组数据,方便人们查看与研究。因此在呈现基因组碱基序列的同时,也结合了注释信息,例如known genes, predicted genes, ESTs, mRNAs, CpG islands, assembly gaps and coverage, chromosomal bands, mouse homologies等等。所以用户既可以用他们提供的数据库里面的数据,也可以上传自己的数据来做研究。围绕着这样的初衷,他们设计
Genome Browser 整合基因组数据和各种注释数据的在线查看系统
Blat 序列比对工具
Table Browser 将文本文件转化为数据库可以识别的文件
Genome Graphs 上传和呈现基因组数据的工具,例如genome-wide SNP association studies, linkage studies 和homozygosity mapping
Gene Sorter 各种形式的呈现基因的表达,同源等信息以及相互关系
Gene Interactions 基因之间的交互关系
In-Silico PCR 查看一对引物在基因组中的位置
VisiGene 查看基因在显微镜下的原位图
LiftOver 基因组版本的转换三、常用案例介绍
1.如何搜索根据位置来快速获得序列
比如:获得chr17:7676091,7676196对应的序列
方法: http://genome.ucsc.edu/cgi-bin/das/hg38/dna?segment=chr17:7676091,7676196
网页会返回 一个xml格式的信息,解析一下即可。
This XML file does not appear to have any style information associated with it. The document tree is shown below.
<DASDNA>
<SEQUENCE id="chr17" start="7676091" stop="7676196" version="1.00">
<DNA length="106">
aggggccaggagggggctggtgcaggggccgccggtgtaggagctgctgg tgcaggggccacggggggagcagcctctggcattctgggagcttcatctg gacctg
</DNA>
</SEQUENCE>
</DASDNA>很明显里面的aggggccaggagggggctggtgcaggggccgccggtgtaggagctgctgg tgcaggggccacggggggagcagcctctggcattctgggagcttcatctg gacctg 就是我们想要的序列啦
hg38可以更换成hg19,dna?segment= 后面可以按照标准格式更换,既可以返回我们想要的序列了。
2.下载数据
首先是NCBI对应UCSC,对应ENSEMBL数据库: GRCh36 (hg18): ENSEMBL release_52. GRCh37 (hg19): ENSEMBL release_59/61/64/68/69/75. GRCh38 (hg38): ENSEMBL release_76/77/78/80/81/82. 可以看到ENSEMBL的版本特别复杂!!!很容易搞混! 但是UCSC的版本就简单了,就hg18,19,38, 常用的是hg19,但是我推荐大家都转为hg38
UCSC里面下载非常方便,只需要根据基因组简称来拼接url即可:
http://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/mm9/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/chromFa.tar.gz或者用shell脚本指定下载的染色体号:
for i in $(seq 1 22) X Y M;
do echo $i;
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;
## 这里也可以用NCBI的:ftp://ftp.ncbi.nih.gov/genomes/M_musculus/ARCHIVE/MGSCv3_Release3/Assembled_Chromosomes/chr前缀
done
gunzip *.gz
for i in $(seq 1 22) X Y M;
do cat chr${i}.fa >> hg19.fasta;
done
rm -fr chr*.fasta- 基因组数据版本转化
染色体区域根据不同的版本(如hg19或者hg38)会有不同的显示,假设我现在有一段hg19上关注的区域(例如chr1:100-10000),我想知道在hg38版本上这段区域的位置,以便于可以更新注释信息,需要怎么做?
三大主流生物信息学数据库运营单位都出了自己的基因组坐标转换,它们分别是 (UCSC的liftOver, NCBI 的 Remap, Ensembl的API),其中Ensembl的API是基于crossmap的,是一个python程序。而ucsc的LiftOver最出名,而且有可执行版本软件可以下载。
- Blat
- 针对DNA序列,BLAT是用来设计寻找95%及以上相似 至少40个碱基的序列。
- 针对蛋白序列,BLAT是用来设计寻找80%及以上相似 至少20个氨基酸的序列。
- 用法:
- 查找mRNA或蛋白在基因组中的位置
- 决定基因外显子的结构
- 显示全长基因的编码区域
- 分离一个物种他自己的EST
- 查找基因家族
- 从其他物种中查找人类基因的同源物
更多参考:https://genome.ucsc.edu/goldenpath/help/hgTracksHelp.html
参考资料
《人类基因组用户指南》
http://www.bio-info-trainee.com/1049.html
http://www.biotrainee.com/thread-29-1-1.html
https://genome.ucsc.edu/goldenpath/help/hgTracksHelp.html#What
3.6 ENCODE
一、简介
人类基因组计划的主要目标是产生人类和主要模式生物(包括大肠杆菌(Saccharomyces cerevisiae)、线虫(Caenorhabditis elegans)、果蝇(Drosophila melanogaster)及小鼠(Mus musculus))的精确基因组序列。研究人员可以免费获取这项研究产生的数据公布,这又促进了人类基因组变异图谱的产生和发展(International HapMap Project)。然而,人们仍然不了解基因组如何编码产生多细胞有机体。这就需要精确阐明基因组上重要功能元件并且描绘出这些原件随着细胞种类及时间变化的动态变化情况。这些原件包括编码蛋白、非编码RNA、重要功能原件(如直接调控基因表达,DNA复制和染色体变异)的调控序列。大肠杆菌拥有较小规模基因组,因此首先被破译。对较为复杂的人、小鼠、果蝇及线虫基因组的破译工作仍然处在起始阶段。因此,美国国立人类基因组研究中心(National Human Genome Research Institute (NHGRI))于2003年启动了ENCODE (Encyclopedia of DNA Elements)计划,该计划的最终目标是描绘出人类基因组的功能元件。在此基础上,对此项计划的扩展,内容包括将对人类基因组破译工作扩展到整个基因组,另外于2007年发起了对模式动物线虫和果蝇基因组破译工作–ENCODE (modENCODE)。
该项目吸引了来自美国、英国、西班牙、日本和新加坡五国32个研究机构的440多名研究人员的参与,经过了9年的努力,研究了147个组织类型,进行了1478次实验,获得并分析了超过15万亿字节的原始数据,确定了400万个基因开关,明确了哪些DNA片段能打开或关闭特定的基因,以及不同类型细胞之间的“开关”存在的差异。证明所谓“垃圾DNA”都是十分有用的基因成分,担任着基因调控重任。证明人体内没有一个DNA片段是无用的。
目前所有数据均全部公开(http://genome.ucsc.edu/ENCODE/ ),并以30篇论文在Nature、Science、Cell、JBC、Genome Biol、Genome Research同时发表(http://www.nature.com/encode )。成为一个互动的百科全书,并可以免费公开获得和利用这些全部的资料和数据。这是迄今最详细的人类基因组分析数据,是对人类生命科学的又一重大贡献。
更多信息见:
ENCODE主页 https://www.encodeproject.org/
modENCODE计划主页 http://www.modencode.org/
nature相关主题资源 http://www.nature.com/nature/focus/encode/index.html
ENCODE and modENCODE Data Listings http://www.ncbi.nlm.nih.gov/projects/geo/info/ENCODE.html
modENCODE 计划相关发表文献 http://blog.modencode.org/category/publications
NIH提供的ENCODE计划相关教程:
- https://www.genome.gov/27553900/encode-tutorials/
- https://www.genome.gov/27562350/encode-workshop-april-2015-keystone-symposia/
- https://www.genome.gov/27561253/encode-workshop-tutorial-october-2014-ashg/
- https://www.genome.gov/27553901/encode-tutorial-may-2013-biology-of-genomes-cshl/
- https://www.genome.gov/27563006/encoderoadmap-epigenomics-tutorial-october-2015-ashg/
- https://www.genome.gov/27555330/encoderoadmap-epigenomics-tutorial-october-2013-ashg/
- https://www.genome.gov/27551933/encoderoadmap-epigenomics-tutorial-nov-2012-ashg/
- http://useast.ensembl.org/info/website/tutorials/encode.html
- https://www.encodeproject.org/tutorials/
- https://www.encodeproject.org/tutorials/encode-meeting-2016/
- https://www.encodeproject.org/tutorials/encode-users-meeting-2015/
二、常见问题
- 6种方式下载ENCODE计划的所有数据
http://www.bio-info-trainee.com/1825.html
所有数据从raw data形式的原始测序数据到比对后的信号文件以及分析好的有意的peaks文件都可以下载。
2.ENCODE计划中enhance和promoter的确定
http://www.biotrainee.com/thread-298-1-1.html
参考资料
http://www.bio-info-trainee.com/1825.html
http://blog.sina.com.cn/s/blog_6a17628d0100vpba.html
3.7 GENCODE
一、介绍
NHGRI( National Human Genome Research Institute)于2003年9月启动了ENCODE计划(Encyclopedia Of DNA Elements),旨在发现人类基因组序列中的功能元件。随后,Sanger被授权启动GENCODE项目,旨在整合基因注释结果的整合,比如基因组每条染色体上面有哪些编码蛋白的基因,哪些假基因,哪些lncRNA的基因,它们坐标是什么,基因上面的外显子内含子坐标是什么,UTR区域坐标是什么。在2013年,GENCODE小组也启动了小鼠基因组的注释信息的整合工作。目前,GENCODE基因信息被ENCODE和1000 Genomes等其他项目使用。
GENCODE 目标
- 提高人类基因注释结构的覆盖度和准确性,特别是蛋白编码的可变剪切突变,非编码位置和假基因等的位置。
- 建立老鼠基因,包含编码蛋白的可变剪切突变,有转录证据的非编码位置,假基因等。
通过比较小鼠注释的数据和人的基因注释结果可以提高注释结果的准确性。注释工作包括人工矫正,不同方法的计算分析和设计实验证明。有争议的位置会通过实验来验证。数据资源可以Ensembl和UCSC等上公开。
Version 26 (October 2016 freeze, GRCh38) - Ensembl90版本的统计数据
- Total No of Genes:58288
- Protein-coding genes: 19836
- Long non-coding RNA genes: 15778
- Small non-coding RNA genes: 7569
- Pseudogenes: 14694
- processed pseudogenes: 10704
- unprocessed pseudogenes: 3469
- unitary pseudogenes: 206
- polymorphic pseudogenes: 63
- pseudogenes: 18
- Immunoglobulin/T-cell receptor gene segments
- protein coding segments: 410
- pseudogenes: 234
- Total No of Transcripts: 200401
- Protein-coding transcripts: 80930
- full length protein-coding: 55406
- partial length protein-coding: 25524
- Nonsense mediated decay transcripts: 14208
- Long non-coding RNA loci transcripts: 27908
- Total No of distinct translations: 60172
- Genes that have more than one distinct translations: 13546
二、数据的下载
FTP地址:ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/
可以下载该数据库的所有资料,而且整理的非常好,自己写脚本很容易处理得到自己想要的信息。
以GENCODE v24为例,在linux系统里面用shell代码即可批量下载所有metadata数据
wget -c -r -np -k -L -A "*metadata*" ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_24/再用代码检查里面的记录数:
ls *gz |while read id;do (echo -n $id;echo -n " " ;zcat $id |wc -l ) ;done我们看看meta data信息的记录数量,这些信息主要是GENCODE与其它主流数据库的对应关系
gencode.v24.metadata.Annotation_remark.gz 40879
gencode.v24.metadata.EntrezGene.gz 170466
gencode.v24.metadata.Exon_supporting_feature.gz 19193542
gencode.v24.metadata.Gene_source.gz 66206
gencode.v24.metadata.HGNC.gz 182831
gencode.v24.metadata.PDB.gz 94547
gencode.v24.metadata.PolyA_feature.gz 84652
gencode.v24.metadata.Pubmed_id.gz 209094
gencode.v24.metadata.RefSeq.gz 75365
gencode.v24.metadata.Selenocysteine.gz 119
gencode.v24.metadata.SwissProt.gz 45067
gencode.v24.metadata.Transcript_source.gz 217202
gencode.v24.metadata.Transcript_supporting_feature.gz 87375
gencode.v24.metadata.TrEMBL.gz 61924还可以下载所有的gtf文件:
wget -c -r -np -nd -k -L -A "*gtf.gz" ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/gtf文件特别重要,具体可参见第二章节数据格式的介绍。
还可以下载参考转录组及参考蛋白组,我这里还是拿hg19举例:
## ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v24lift37.transcripts.fa.gz
## ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v24lift37.lncRNA_transcripts.fa.gz
## ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v24lift37.pc_transcripts.fa.gz其实你有gtf文件,也可以直接从参考基因组序列里面提取这个参考转录组及参考蛋白组,通常是gtf2fasta,随便搜索一下,一大堆方法。
三、常见问题
- 根据gtf格式的基因注释文件得到人所有基因的染色体坐标
http://www.biotrainee.com/thread-472-1-1.html
2.常用的一些提取信息的命令
获得基因名列表
``awk '{if($3=="gene"){print $0}}' gencode.gtf``
获得所有的"protein-coding transcript"行
``awk '{if($3=="transcript" && $20=="\"protein_coding\";"){print $0}}' gencode.gtf``
获得手动注释级别为1或2的注释结果
``awk '{if($0~"level (1|2);"){print $0}}' gencode.gtf``参考资料
http://www.bio-info-trainee.com/1781.html
http://www.gencodegenes.org/about.html
The GENCODE v7 catalog of human long noncoding RNAs, 链接 http://genome.cshlp.org/content/22/9/1775.full
3.8 TCGA
一、简介
肿瘤基因组图谱 (The Cancer Genome Atlas,TCGA)计划由美国 National Cancer Institute(NCI)和National Human Genome Research Institute(NHGRI)于2006年联合启动的项目。目前收录了来自11000个病人,33个癌症的数据,2.5P的数据量。 但是TCGA只对授权的用户开放Level1-Level3数据访问的权限,而普通用户只能访问Level3的分析结果。即TCGA数据库的普通用户无法用Level1的数据进行个性化的高级分析。同时,这些用户也不能有效结合重要的临床信息进行数据的深入挖掘,严重限制用户对数据的有效利用。
收录的癌症类型,详见:https://cancergenome.nih.gov/cancersselected
Platform Design
- Applied Biosystems Sequence data
- Agilent 244K Custom Gene Expression
- Agilent SurePrint G3 Human CGH Microarray Kit
- Affymetrix Genome-Wide Human SNP Array
- Agilent 8 x 15K Human miRNA-specific microarray
- Illumina Genome Analyzer DNA Sequencing 等等
更多阅读:https://cancergenome.nih.gov/abouttcga/aboutdata/platformdesign
TCGA数据类型
数据类型包括:Clinical Data,Images,Microsatellite Instability (MSI),DNA Sequencing,miRNA Sequencing, Protein Expression,mRNA Sequencing,Total RNA Sequencing ,Array-based Expression ,DNA Methylation, Copy Number
更详尽的关于数据类型和数据等级,参见:https://cancergenome.nih.gov/abouttcga/aboutdata/datalevelstypes
癌症样本组织处理
癌症病人自愿捐赠肿瘤组织及正常组织样本,由人类癌症生物标本核心资源库承担癌症组织标本和正常组织标本的采集、处理和分配工作。
组织样本经过严格标准处理(处理标准根据不同后续分析类型而异,具体标准请参见),确保质量可以用于进一步分析及测序,并由相关中心采用高通量测序技术进行基因和基因组排序。
获得的临床资料中,可以识别病人身份的信息去掉。
TCGA个部门分工
TCGA 基因组分析中心(GCC)比对肿瘤和正常组织,寻找异常的基因重组现象。
高通量测序中心(GSC)分析与各癌症或者亚型相关的基因突变、扩增或者缺失。
资料分析中心(GDAC)进行资料的整理、汇总、并提供图表报告给全体研究团队。
资料分享
资料综合中心(DCC)集中处理各个团队产生的资料,定期公开于网络上供全世界研究人员利用。
提供公开的资料下载网站入口以方便进行资料搜索和下载
二、数据下载
虽然在TCGA中直接下载数据的方法较为繁琐,但是有多个网站提供TCGA数据(包括表达和临床等)完善的整理:GDAC, Cancer Browser和cBioportal是其中整理最为完整和可靠的。GDAC由美国MIT和Harvard共建的Broadinstitute运行,UCSC运行着Cancer Browser 和Xena, cBioportal由MemorialSloan-Kettering Cancer Cente建立,提供较为完善的TCGA数据为基础的各类信息检索服务。
下载的数据分为两个权限:
- 公开的数据
这部分数据不涉及个人信息,下载这部分数据不需要用户认证,包括的数据
- De-identified clinical and demographic data
- Gene expression data
- Copy number alterations in regions of the genome
- Epigenetic data
- Summaries of data compiled across individuals
- Anonymized single amplicon DNA sequence data
- 受控的数据
因为这部分信息设计到个人信息,所有需要用户申请,包括的数据:
- Primary sequence data (BAM and FASTQ files)
- SNP6 array level 1 and level 2 data
- Exon array level 1 and level 2 data
- VCFs
- Certain information in MAFs
下载途径
- GDC
自2016年7月15日起,TCGA(The Cancer Genomic Atlas) DATA PORTAL不再提供数据服务,所有数据将转入GDC(Genomic Data Commons) DATA PORTAL。 GDC网站下载TCGA数据,图形界面,操作简单。
GDC提供两种数据下载方式:
(1)对于少量数据,在购物车内点击download,选择cart可以直接下载购物车内的数据
(2)对于大量数据,从购物车中直接下载易出现错误。我们可以点击download下的manifest,然后利用GDC Transfer Tool (gdc-client),在Terminal内输入如下命令进行批量下载: ./gdc-client download -m manifest_xxx.txt
更多阅读:http://www.biotrainee.com/thread-821-1-1.html
- gdac和firehose
网站是:https://gdac.broadinstitute.org/
客户端工具是firehose_get ,https://confluence.broadinstitute.org/display/GDAC/Download
这里的数据也来源于 portal.gdc.cancer.gov,经过了简单的合并,将每种癌症相同类型的数据合并到了一个文件中(例如443个胃癌样本的RNA表达量数据都合并到了一个文件中,非常适合用R进行后续的分析)
更多阅读:
http://www.biotrainee.com/thread-822-1-1.html
http://www.biotrainee.com/thread-822-1-2.html
- cgdsR和cbioportal
cbioportal地址是:http://www.cbioportal.org/
他们给网站做了一个R包的API为cgdsR,整合和简化了包括TCGA,ICGC以及GEO等多个癌症基因组数据库的内容,提供友好可视化的界面,可供下载。
主要展示基因的somatic 突变谱,拷贝数变化,mRNA&miRNA表达量变化,DNA甲基化以及蛋白质表达的情况,并结合患者的临床资料,展示了KM生存曲线。
更多阅读:http://www.biotrainee.com/thread-824-1-3.html
- Synapse
Synapse是需要注册的,但是是免费注册的,很简单,用谷歌账户注册即可。
https://www.synapse.org/#!Synapse:syn300013
这里面存放的就是一系列TCGA大文章的数据,一些人整理好的,所以非常方便的可以使用! 比如,我们可以获取 Lung Squamous Cell Carcinoma的生存分析数据
https://www.synapse.org/#!Synapse:syn1446127/version/3
三、常用分析工具
- cBioPortal(cBio Cancer Genomics Portal)
是一个基于TCGA数据库,进行可视化分析的网页。
官网: http://cbioportal.org
首先进入这个网页(http://cbioportal.org),然后可以看到下面这个界面,首先选择你想要分析的数据库和具体的数据
接着勾选你要分析的数据到底都是啥,主要可以分析的是MUT(Mutation,突变),CNA(Copy Number Alterations,拷贝数变化),EXP(mRNA Expression,mRNA表达)和PORT/RPPA(Protein/ phosphoprotein level,蛋白表达或磷酸化变化)。但要注意的是,并不是所有数据都具备这四个选项,大多数只有MUT和CNA这两组数据,有些具有EXP数据和PORT数据。接着,要选择你要研究的基因,有一个下拉菜单可以给你参考,比如会有类似信号通路上的明星分子集合这类,你可以按照需要选择。当然,也可以自己输入基因名
确认后就可以进入结果页面了,主要是显示样本中较为直观的变化,比如突变、缺失、RNA表达、磷酸化变化等等。
- 用TANRIC来探索癌症中lncRNA功能
更多阅读:http://www.biotrainee.com/thread-999-1-2.html
- TCGA2BED-可以从TCGA数据库提取数据成bed格式
官网:http://bioinformatics.mdanderson.org/main/TANRIC:Overview
更多阅读:http://www.biotrainee.com/thread-1056-1-1.html
- TCGA可视化数据库GEPIA
官网:http://gepia.cancer-pku.cn/index.html
这个数据库可以分析有什么功能呢?
给一个基因,告诉你在所有肿瘤组织里面的表达情况,同时还展示其在癌和癌旁的表达
给一个基因,自动做生存分析
给一个基因,告诉你他的共表达基因,或者叫表达模式相似的基因
给两个基因,告诉你他在特定组织的相关性
可以做编码基因,也可以做非编码基因
- TCGA生存分析oncolnc
官网:http://www.oncolnc.org/
这是一个整合了TCGA的各种RNA数据和患者临床数据,提供生存分析的网站,灰常简单好用。
- 基于TCGA的蛋白芯片分析神器TCPA
官网:http://www.tcpaportal.org/tcpa/
更多阅读:http://www.biotrainee.com/thread-1293-1-1.html
- UCSC的cancer genome browser探索TCGA的level3数据
可以对任何癌种,根据任何临床指标进行分sub-group之后进行任何形式的生存分析,比较分析,还有相关分析。
更多阅读: http://www.biotrainee.com/thread-1086-1-1.html
- Immunophenogram
网站是:https://tcia.at/home
TCGA的数据挖掘大文章类型,从细胞群里里面区分各种免疫细胞
- 基于TCGA的甲基化神器mexpress
官网:http://mexpress.be/
整合了TCGA中的DNA甲基化,表达量及临床数据,主要用来探索甲基化,基因表达和临床表型之间的关联
- oncomine
Oncomine是目前最大的癌症基因芯片数据库 更多阅读:http://www.biotrainee.com/thread-1242-1-1.html
参考资料
http://www.biotrainee.com/thread-1080-1-1.html
http://www.biotrainee.com/thread-306-1-1.html
http://www.biotrainee.com/thread-307-1-1.html
http://www.biotrainee.com/thread-827-1-1.html
http://www.biotrainee.com/thread-1290-1-1.html
http://paper.dxy.cn/article/511878
https://cancergenome.nih.gov/abouttcga/overview
##1000 GENOME
一、简介
千人基因组计划(1000 Genomes,http://www.internationalgenome.org/ )于2008-2015年开启的对人的基因组进行测序一个项目,目的是建立人类突变和分型的共同数据库。虽然这个项目已经结束了,EMBL-EBI的数据中心仍然获得了Wellcome Trust的基金资助来维护和扩充这个数据库。 IGSR(International Genome Sample Resource)想实现的目标:
- 保证公众能访问和使用1000 Genomes的数据
- 补充已发表的基因组其他信息
- 向1000 Genomes持续增加新的基因组数据
项目完成的三个阶段
| 阶段 | 目标 | 深度 | 策略 | 状态 |
|---|---|---|---|---|
| 1-low coverage | Assess strategy of sharing data across samples | 2-4X | 180个样本全基因组测序 | 2008年9月完成 |
| 2-trios | Assess coverage and platforms and centres | 20-60X | 2个母亲-父亲-孩子的家系测序 | 2008年10月完成 |
| 3-gene regions | Assess methods for gene-region-capture | 50X | 900个样本1000个基因 | 2009年6月完成 |
5个大的人种(亚洲人、欧洲人等),25个亚人种。目前,新版共有NA编号开头的1182个人,HG开头的1768个人。
它的官方网站是:有一个ppt讲得很清楚如何通过官网做的data portal来下载数据:https://www.genome.gov/pages/research/der/ichg-1000genomestutorial/how_to_access_the_data.pdf
二、数据查询与下载
查询数据
千人基因组计划 – 基因组浏览器: http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/
查询某个SNP的信息
http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=rs35761398
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2501432
http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2502992在千人基因组计划里面看一个rs就能看到各种人群信息: http://browser.1000genomes.org/Homo_sapiens/Variation/Population?r=1:24201420-24202420;v=rs2501432;vdb=variation;vf=1849472 这些人群信息,可以画一个网路图! 只需要变化rs ID号即可,当然并不是所有的rs ID号都在千人基因组计划里面有显示的。
下载数据
下载地址:
ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/
ftp://ftp.sanger.ac.uk/pub/1000genomes/
ftp://ftp.ebi.ac.uk/pub/databases/1000genomes/
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp直接看最新版的数据,共有NA编号开头的1182个人,HG开头的1768个人! ftp://ftp.ncbi.nlm.nih.gov/1000genomes/ftp/phase3/data/
也可以按照人种来查看这些数据:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/data/
每个人的目录下面都有 四个数据文件夹
Oct 01 2014 00:00 Directory alignment
Oct 01 2014 00:00 Directory exome_alignment
Oct 01 2014 00:00 Directory high_coverage_alignment
Oct 01 2014 00:00 Directory sequence_read这些数据实在是太丰富了!
也可以直接看最新版的vcf文件,记录了这两千多人的所有变异位点信息! 可以直接看到所有的位点,具体到每个人在该位点是否变异! ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/
不过它的基因型信息是通过MVNcall+SHAPEIT这个程序call出来的,具体原理见:http://www.ncbi.nlm.nih.gov/pubmed/23093610
而且网站还提供一些教程:ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/working/
表达相关的数据
1000G项目中比较重要的表达数据:
- 465个个体(包括种群:CEU, TSI, GBR, FIN, YRI)的RNAseq数据(mRNA和miRNA)
http://www.geuvadis.org
http://www.ebi.ac.uk/arrayexpress/experiments/E-GEUV-1/samples.html
http://www.ebi.ac.uk/arrayexpress/experiments/E-GEUV-2/samples.html
- 60个CEU个个体RNAseq
http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-197
- 800 HapMap个体的表达芯片
http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-198
http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-264
- 69个YRI个体的RNAseq数据
http://www.ebi.ac.uk/arrayexpress/experiments/E-GEOD-19480
三、讨论
- 1000G中的突变也在dbSNP中吗?
1000G中的SNP和插入缺失突变都提交到了dbSNP中,更长的结构突变被提交到了DGVa。 1000G的vcf文件中有一个ID列,对应的就是dbSNP的rs ID。 因为方法改进,phase 3为代表最终的结果
参考资料
http://www.bio-info-trainee.com/1841.html
http://www.internationalgenome.org/about
http://www.internationalgenome.org/category/dbsnp